Plasmazellmyelom
Das Plasmazellmyelom, auch Multiples Myelom oder Plasmozytom genannt, ist eine reife B-Zell-Neoplasie, die durch eine multifokale Proliferation neoplastischer Plasmazellen im Knochenmark gekennzeichnet ist. Diese Zellen produzieren monoklonale Antikörper, die als Paraproteine bekannt sind und als Marker für die Krankheitsaktivität dienen. Die Erkrankung führt zu Störungen in der Blutproduktion, der Knochensubstanz, der Nierenfunktion und des Calciumstoffwechsels. Diagnostisch werden neben der Infiltration des Knochenmarks und der Höhe des monoklonalen Proteins in Serum und Urin auch Endorganschäden und genetische Faktoren berücksichtigt, wobei zur Prognoseabschätzung das Revidierte Internationale Staging System (R-ISS) verwendet wird.
Wissenschaftlicher Hintergrund
Das multiple Myelom (MM) zählt zu den reifen B-Zell-Neoplasien. Es beruht auf einer Knochenmark-basierten, multifokalen Proliferation von neoplastischen Plasmazellen (WHO 2017). Plasmazellen stellen die Antikörper-produzierenden Effektorzellen der B-Zell-Reihe dar, und auch die neoplastischen Plasmazellen produzieren charakteristischerweise monoklonale komplette oder inkomplette Antikörper. Diese sogenannten Paraproteine oder M-Proteine sind im Serum und/oder Urin nachweisbar und eignen sich als Verlaufsparameter für die Krankheitsaktivität. Durch die fehlerhaft im Knochenmark proliferierenden Myelomzellen, die Sekretion der Paraproteine sowie zusätzliche, von den neoplastischen Zellen abgesonderte Botenstoffe, kommt es zu Störungen der Blutproduktion, der Knochensubstanz, der Nierenfunktion und des Calciumstoffwechsels. Die Einordnung in das jeweilige Krankheitsstadium ist komplex und wird über den Infiltrationsgrad des Knochenmarks, die Höhe des monoklonalen Proteins in Serum und Urin und Endorganschäden (=Calciumstoffwechsel, Renale Schäden, Anämie, Bone), die sogenannten SLiM-CRAB-Kriterien oder auch Myelom-definierende Biomarker, definiert.
Klinisch und auch genetisch ist das Multiple Myelom sehr heterogen. Vom Vollbild des MM abzugrenzen sind dessen klinische Vorstufen, die Monoklonale Gammopathie unklarer Signifikanz (MGUS) und das schwelende (smouldering) Myelom.
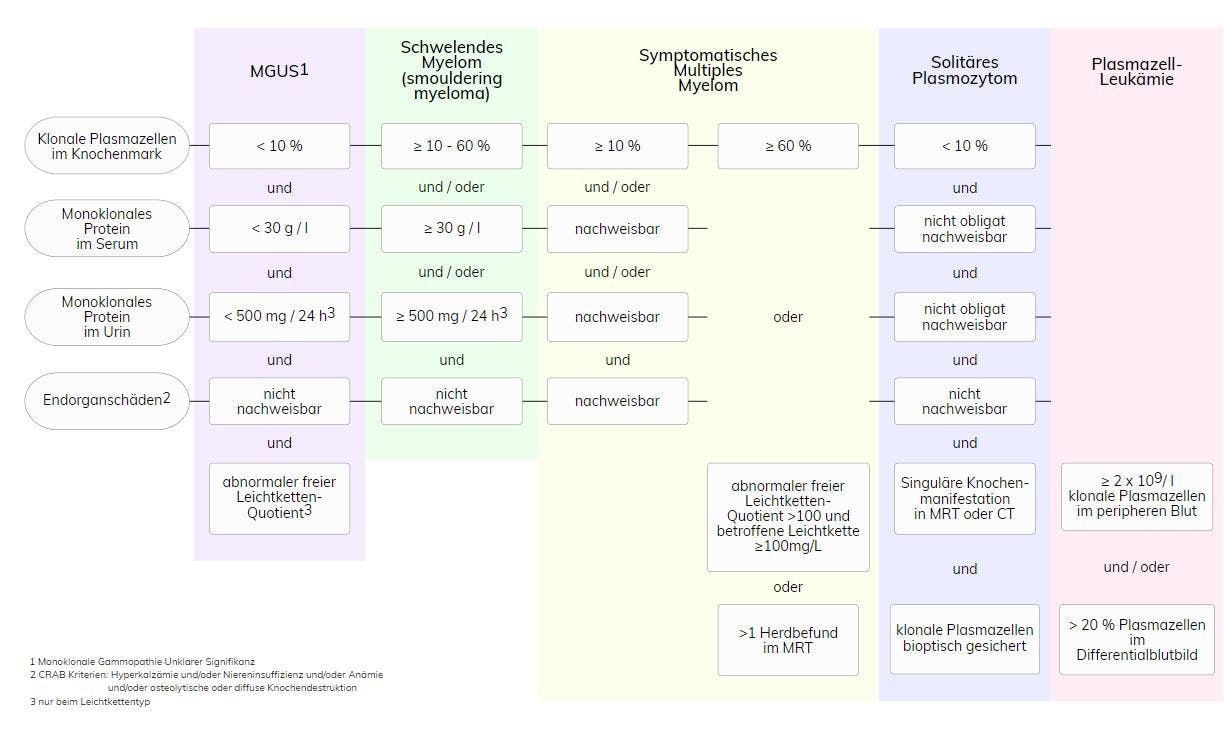
Als Vorläuferläsion kann die Monoklonale Gammopathie unklarer Signifikanz (MGUS) angesehen werden. Sie beruht auf einer monoklonalen Proliferation von Plasmazellen, muss jedoch nicht in eine maligne Erkrankung übergehen. Sie ist definiert als laborchemischer Nachweis kompletter oder inkompletter monoklonaler Immunglobuline im Serum von Personen ohne klinische Symptomatik. Ein Paraprotein sollte aber immer Anlass zu einer umfassenden Diagnostik geben, um eine potentiell behandelbare Paraprotein-vermittelte Erkrankung möglichst früh zu erkennen. Das Risiko einer Progression in eine maligne Erkrankung oder Amyloidose kann anhand der Parameter Höhe des monoklonalen Proteins ≥15g/l, abnormer kappa/lambda-Leichtketten-Quotient im Serum, nicht-IgG-MGUS abgeschätzt werden. Bei Vorliegen aller drei Risikofaktoren liegt die Progressionswahrscheinlichkeit nach 20 Jahren bei über 50% (Hochrisiko-MGUS), bei Fehlen aller Risikofaktoren lediglich bei 5% (Niedrigrisiko-MGUS).
Bei dem MM wird zur besseren Abschätzung der Prognose aktuell das Revidierte Internationale Staging System (revised ISS) der International Myeloma Working Group (IMWG) verwendet, das betroffene Patienten in 3 prognostische Subgruppen einteilt (Stadium I-III, 5-Jahres-Überlebensrate 82%/62%/40%). Die Stadieneinteilung nach Salmon und Durie wird heute nicht mehr verwendet. In den R-ISS fließen zytogenetische Befunde mit ein.
Der diagnostische Algorithmus bei V.a. MM ist komplex und bedarf einer Reihe serologischer, immunologischer, zytologischer, genetischer und bildgebender Untersuchungen.
Stadium | Revised ISS |
| Stadium I | beta 2-Mikroglobulin ≤3,5 mg/l und Albumin ≥3,5 g/dl und Zytogenetik Standardrisiko und LDH ≤ oberer Normwert |
| Stadium II | weder Stadium I noch Stadium III |
| Stadium III | beta 2-Mikroglobulin ≥5,5 mg/l und Zytogenetik Hochrisiko oder LDH > oberer Normwert |
Hochrisiko-Zytogenetik: del17p, t(4;14), t(14;16)
Standard-Risiko: alle anderen zytogenetischen Aberrationen
Diagnostik
Der initiale Untersuchungsschritt bei V.a. MM ist die Knochenmarkaspiration mit –biopsie. Die zytomorphologische/histopathologische Untersuchung auf Plasmazellinfiltrate dient in erster Linie der Abgrenzung zum MGUS und Smouldering Myeloma (SMM). Mittels Immunhistochemie (IHC) bzw. Immunphänotypisierung kann ebenfalls einerseits eine Quantifizierung, andererseits der Nachweis einer Monoklonalität erfolgen. Mittels IHC für aberrant exprimierte Proteine kann die neoplastische Natur der Plasmazellinfiltrate bestätigt werden (CD56, KIT). Gelegentlich kann eine vermehrte MYC-Expression und in Fällen mit einem t(11;14)(IGH-CCND1)-Rearrangement oder Hyperdiploidie eine CyclinD1-Expression nachgewiesen werden.
Durchflusszytometrisch können Myelomzellen im peripheren Blut ebenfalls anhand ihres Immunphänotyps erfassst werden. So exprimieren sie üblicherweise neben CD138 auch CD38, CD45 ist niedrig oder negativ. Ebenso CD19 (in >95% negativ), CD27 und CD81 sind negativ oder schwach exprimiert. MM-Zellen exprimieren üblicherweise kein membranständiges Ig und monotypisches zytoplasmatisches Ig (Leichtkettenrestriktion). Aberrant exprimierte Antigene umfassen CD56, CD200, CD28, KIT, CD20, CD52, CD10. Auch für die Verlaufsdiagnostik nach Therapie (minimale Resterkrankung, MRD) etabliert sich die Immunphänotypisierung zunehmend.
Zytogenetische Aberrationen beim Multiplen Myelom zählen zu den wiederkehrenden Veränderungen beim MM, die derzeit die stärkste prognostische und prädiktive Bedeutung und daher zum Teil auch Eingang in den ISS gefunden haben. Allerdings werden – bedingt durch die geringe Proliferationsaktivität – die aberranten Plasmazellen in einer klassischen Chromosomenanalyse meist nicht erfasst. Daher kommt der Fluoreszenz-in situ-Hybridisierung an zuvor angereicherten, CD138-positiven Zellen eine besondere Bedeutung zu. Hierdurch können in bis zu 90% aller MM chromosomale Veränderungen nachgewiesen werden. Dabei werden primäre (early event) und sekundäre (progression event) genetische Aberrationen unterschieden. Als primäre Aberrationen gelten Trisomien und IGH-Translokationen, also chromosomale Rearrangements unter Einbezug des IGH-Locus auf Chromosom 14.
Diese beiden Ereignisse schließen sich in bis zu 90% der Myelome aus, so dass folgende Gruppen unterschieden werden:
- hyperdiploide Myelome, etwa 45% aller Myelome, mit multiplen Trisomien der Chromosomen 3, 5, 7, 9, 11, 15, 19, 21 und eher günstiger Prognose
- nicht-hyperdiploide Myelome,etwa 40%, mit Translokationen unter Beteiligung der Region 14q32; am häufigsten: t(11;14), t(4;14), t(14;16), t(6;14), t(12;14) und t(14;20) mit weiteren zytogenetischen Aberrationen und variabler Prognose
- andere (unklassifiziert), mit Trisomien und IGH-Translokation, etwa 15% aller MM
Zu den sekundären genetischen Aberrationen zählen vor allem Deletionen (1p, 13q, 17p), Zugewinne von 1q und 11q und MYC-Rearrangements. Häufig finden sich komplexe zytogenetische Befunde beim MM. Eine del17p, del1p und eine ampl1q21 sind mit einer ungünstigen Prognose verbunden. Um der Komplexizität der genetischen Landschaft beim MM Rechnung zu tragen, wurde ein kombinierter zytogenetischer Risikoscore ((t(4;14), del(17p), Trisomie 5, Trisomie 21, 1q Zugewinn, del(1p32)) vorgeschlagen, der eine höhere Vorhersage-Genauigkeit als der R-ISS erreicht. Der klinische Stellenwert dieses Scores ist noch nicht geklärt.
Molekulargenetische Untersuchungen gewinnen – sowohl prognostisch als auch prädiktiv – zunehmend an Bedeutung. Dabei sind überwiegend Komponenten des Ras-Raf/MEK/ERK-Signalwegs betroffen. Aktivierende Varianten wurden in KRAS (23%), NRAS (20%) und BRAF gefunden, die jedoch praktisch nie gemeinsam auftreten. Bei 8% der Patienten wurden Varianten im TP53-Gen identifiziert, die mit einer del(17p) und einer ungünstigen Prognose assoziiert sind. Auch Varianten in einer Vielzahl weiterer Gene konnten identifiziert werden, deren klinischer Stellenwert noch nicht abschließend geklärt ist (TRAF3, PRDM1, CYLD, RB1, IRF4, EGR1, MAX, HIST1H1E und ACTG1).
Auch epigenetischen Veränderungen, miRNAs und dem Tumor-Mikroenvironment wird eine zunehmende Bedeutung in der Pathogenese und Aufrechterhaltung der Erkrankung beigemessen. Diese und weitere genetische Aberrationen und Genexpressionsprofile sind Gegenstand aktueller Forschung.
Die Therapie des multiplen Myeloms ist komplex. Aktuell kommen neben klassischen Chemotherapeutika und Glucocorticoiden vor allem Proteasominhibitoren (z.B. Bortezomib, Carfilzomib, Pomalidomid) und Immunmodulatoren (z.B. Lenalidomid, Thalidomid) aber auch HDAC-Inhibitoren (Panobinostat) und monoklonale Antikörper (z.B. Daratumumab, Elotuzumab) zum Einsatz. Die hochdosierte Chemotherapie mit nachfolgender autologer Stammzelltransplantation hat nach wie vor einen festen Stellenwert in der Erstlinientherapie des symptomatischen MM (SLiM-CRAB-Kriterien). Wenn immer möglich, sollten Patienten im Rahmen klinischer Studien behandelt werden. Eine Vielzahl weiterer neuer Substanzen, unter anderem zielgerichtete Substanzen wie BRAF-Inhibitoren, befindet sich aktuell in Prüfung.
Tab.: Basis-Labordiagnostik bei MM (nach Onkopedia-Empfehlungen von 2018)
| Blutbild | Leukozyten mit Differenzialblutbild |
| Labor | Elektrolyte (Natrium, Kalium, korrigiertes Kalzium) Nierenretentionsparameter (Kreatinin incl. berechneter GFR, Harnstoff) Gesamteiweiß und Albumin im Serum Serumprotein-Elektrophorese (SPE) mit Bestimmung des M-Gradienten Immunfixations-Elektrophorese im Serum und Urin Immunglobuline (IgG, IgA, IgM) im Serum, quantitativ freie Kappa- und Lambda-Leichtketten im Serum quantitativ incl. Berechnung des Quotienten 24 h-Sammelurin zur Quantifizierung der Eiweißausscheidung LDH, GPT beta2-Mikroglobulin im Serum |
| Knochenmark | Aspirat: Zytomorphologie Zytogenetik mittels Fluoreszenz in-situ Hybridisierung (FISH) mindestens del(17p), t(4;14), t(14;16) fakultativ (14;20) und ampl 1q21 Biopsie: Histologie mit Immunphänotypisierung |
Perrot et al. 2019, JCO 37:1657/ Perrot et al. 2018, Blood 132:2456 / Wörmann et al. 2018, Onkopedia Leitlinien der DGHO / Kumar et al. 2017, Nature Rev 3:1 / Swerdlow, Campo, Harris, Jaffe, Pileri, Stein, Thiele (Eds), 2017, WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition), Chapter 13:241 / Rajan et al. 2015, Blood Canc Journal 5:1 / Rajkumar et al. 2014, Lanc Oncology 15:e538
letzte Aktualisierung: 3.11.2023