Hyperoxalurie
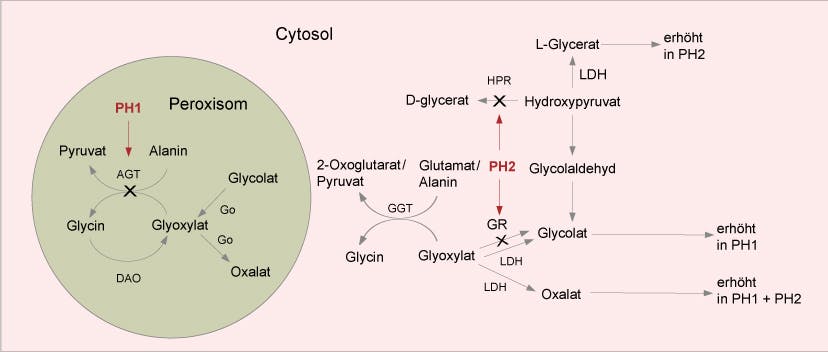
Die primäre Hyperoxalurie Typ 1 ist eine seltene, autosomal-rezessive Erkrankung, die zu Nierenversagen führen kann. Sie wird durch pathogene Veränderungen im AGXT-Gen verursacht, das für das Enzym Alanin-Glyoxylat-Aminotransferase (AGT) codiert. Bei Fehlen von AGT wird Glyoxylat in Oxalat umgewandelt, das sich in der Niere und anderen Organen ablagert. Die Diagnose wird oft innerhalb der ersten sechs Lebensmonate gestellt. Es gibt auch andere Formen der primären Hyperoxalurie, Typ 2 und Typ 3, die durch Defizienzen anderer Enzyme verursacht werden und seltener und weniger schwerwiegend sind.
Wissenschaftlicher Hintergrund
Die primäre Hyperoxalurie Typ 1 ist eine seltene autosomal-rezessive Erkrankung mit einer populationsabhängigen Inzidenz von 1:100.000 – 1:250.000. Die Erkrankung tritt am häufigsten bei Arabern, in Tunesien und im Iran auf. Sie manifestiert sich zwischen der Neugeborenenzeit und dem späten Erwachsenenalter durch Nephrolithiasis und Nephrokalzinose infolge der Ablagerung von Calciumoxalat in den Harnwegen und dem Nierenparenchym und führt unbehandelt zum terminalen Nierenversagen. Bei 10% der Patienten wird die Diagnose innerhalb der ersten sechs Lebensmonate anhand einer Gedeihstörung, Nephrokalzinose, Anämie und einer metabolischen Azidose gestellt. Es handelt sich um die häufigste und schwerste Form der primären Hyperoxalurie (PH). Ursache ist die Defizienz des peroxisomalen Enzyms Alanin-Glyoxylat-Aminotransferase (AGT), das die Umwandlung von Glyoxylat zu Glycin in der Leber katalysiert. Beim Fehlen von AGT wird Glyoxylat in Oxalat umgewandelt, das sich als unlösliches Calciumsalz in der Niere und anderen Organen ablagert. Labordiagnostisch ergeben sich Hinweise aus dem Verhältnis von Oxalsäure zu Kreatinin im Urin, der Oxalsäure-Konzentration im Plasma und der Aktivität von AGT in einer Leberbiopsie.
Ursache der PH1 sind pathogene Veränderungen im AGXT-Gen, welches aus 11 Exons besteht und in 2 normalen Allelzuständen vorkommt:
- Major-Allel (Frequenz von etwa 80% bei Kaukasiern)
- Minor-Allel (Frequenz von etwa 20% bei Kaukasiern, 2% bei Japanern und 3% in der schwarzen Bevölkerung Südafrikas)
Der Minor-Allel Haplotyp weist neben anderen polymorphen Varianten zwei Aminosäuresubstitutionen auf (p.Pro11Leu und p.Ile340Met). 50% aller Betroffenen weisen zusätzlich zu einer ursächlichen Variante mindestens ein Minor-Allel auf. Bisher sind über 200 verschiedene pathogene Veränderungen beschrieben, etwa die Hälfte davon sind Missense-Varianten, daneben aber auch translationale Stoppmutationen und größere genomische Rearrangements. In ca. 80% der PH-Patienten können AGXT-Varianten nachgewiesen werden. Das Genprodukt AGT wird in der Leber synthetisiert und ist in den Peroxisomen lokalisiert. Eine häufige Variante p.Gly170Arg kommt in 25-40% aller PH1-Allele auf dem genetischen Hintergrund des Minor-Allels vor. Dadurch werden 90% der Aminotransferase, die normale katalytische Aktivität aufweist, in die Mitochondrien anstatt in die Peroxisomen fehlgeleitet, wobei sie dort keinen Kontakt mit ihrem Substrat haben. 50% aller Patienten weisen ein komplettes Fehlen von AGT auf, in 20% wird ein funktionell inaktives Enzym synthetisiert, und in seltenen Fällen wird ein Enzym mit verminderter Aktivität produziert, das mit einer milden Verlaufsform assoziiert ist. Heterozygote Anlageträger sind asymptomatisch.
Differentialdiagnostisch ist die Primäre Hyperoxalurie Typ 2 (PH2) zu nennen, die durch eine Defizienz des Enzyms Glyoxylat-Reduktase (GR) verursacht wird und seltener und weniger schwerwiegend als PH1 ist. GR katalysiert die Reduktion von Glyoxylat und Hydroxypyruvat und wird vom GRHPR-Gen codiert. Ursächliche Varianten im GRHPR-Gen wurden in bis zu 10% der genetisch untersuchten PH-Patienten nachgewiesen.
Primäre Hyperoxalurie Typ 3 (PH3) ist bei 5% der Patienten durch erhöhtes Oxalat und Glykolat gekennzeichnet, wobei die Enzymaktivitäten von AGT und GR normal sind. Die Verlaufsform ist weniger gravierend als bei PH1 und PH2. Ursache für PH3 sind ursächliche Varianten im HOGA1-Gen, das für 2-Keto-4-Hydroxy-Glutarat-Aldolase codiert. Pathogene Veränderungen wurden in etwa 10% der positiven PH-Fälle detektiert.
Cai et al. 2018, BMC Nephrol 20:224 / Pelle et al. 2017, J Nephrol 30:219 / Hulton 2016, Int J Surg 36:649 / Ben-Shalom et Frishberg 2015, Pediatr Nephrol 30:1781 / Hopp et al. 2015, J Am Soc Nephrol 26:2559 / Pey at al. 2013, Biomed Res Int 2013:687658 / Williams et al. 2009, Hum Mutat 30:910 / Williams and Rumsby 2007, Clin Chemistry 53:1216 / Coulter-Mackie et Lian 2006, Mol Genet Metab 89:349 / Danpure et al. 2005, Am J Nephrol 25:303 / Monico et al. 2005, Kidney Int 67:1704 / Coulter-Mackie et Rumsby 2004, Mol Genet Metab 83:38 / Pirulli et al. 2003, J Nephrol 16:297 / Lumb et Danpure 2000, J Biol Chem 275:36415 / Amoroso et al. 1999, Hum Genet 104:441
letzte Aktualisierung: 31.3.2024