Noonan Syndrom
Das Noonan-Syndrom ist eine autosomal-dominant vererbte Erkrankung, die 1 von 1.000 bis 2.500 Lebendgeburten betrifft. Es zeichnet sich durch ein breites Spektrum an Symptomen aus, darunter Gesichtsdysmorphien, Kleinwuchs, Brustdeformationen und Herzfehler. Die Hauptursache des Syndroms ist in etwa 50% der Fälle auf pathogene Varianten im PTPN11-Gen zurückzuführen, das für eine Protein-Tyrosin-Phosphatase codiert. Darüber hinaus wurden pathogene Varianten in weiteren Genen identifiziert, die mit dem Noonan-Syndrom in Verbindung stehen und häufig Herzprobleme verursachen.
Wissenschaftlicher Hintergrund
Beim Noonan-Syndrom handelt es sich um eine autosomal-dominant vererbte Erkrankung, deren Häufigkeit mit 1:1.000-2.500 Lebendgeburten angegeben wird. Das variable Symptomenspektrum umfasst faziale Dysmorphien (breite Stirn, Hypertelorismus, Ohrentiefstand, Lidachsenverlauf nach außen unten), proportionierten Kleinwuchs, Pterygium colli, Brustdeformationen, Kryptorchismus, mentale Retardierung, eine leichte Blutungsneigung sowie Herzfehler, meist in Form einer Pulmonalstenose oder einer hypertrophen Kardiomyopathie. Das PTPN11-Gen, das für eine zytoplasmatische Protein-Tyrosin-Phosphatase (SHP-2) codiert, ist das hauptsächlich bei Noonan-Syndrom betroffene Gen. Pathogene Varianten im PTPN11-Gen, die fast ausschließlich zu Aminosäureaustauschen führen, sind die molekulare Ursache in etwa 50% aller bisher untersuchten Noonan-Patienten.
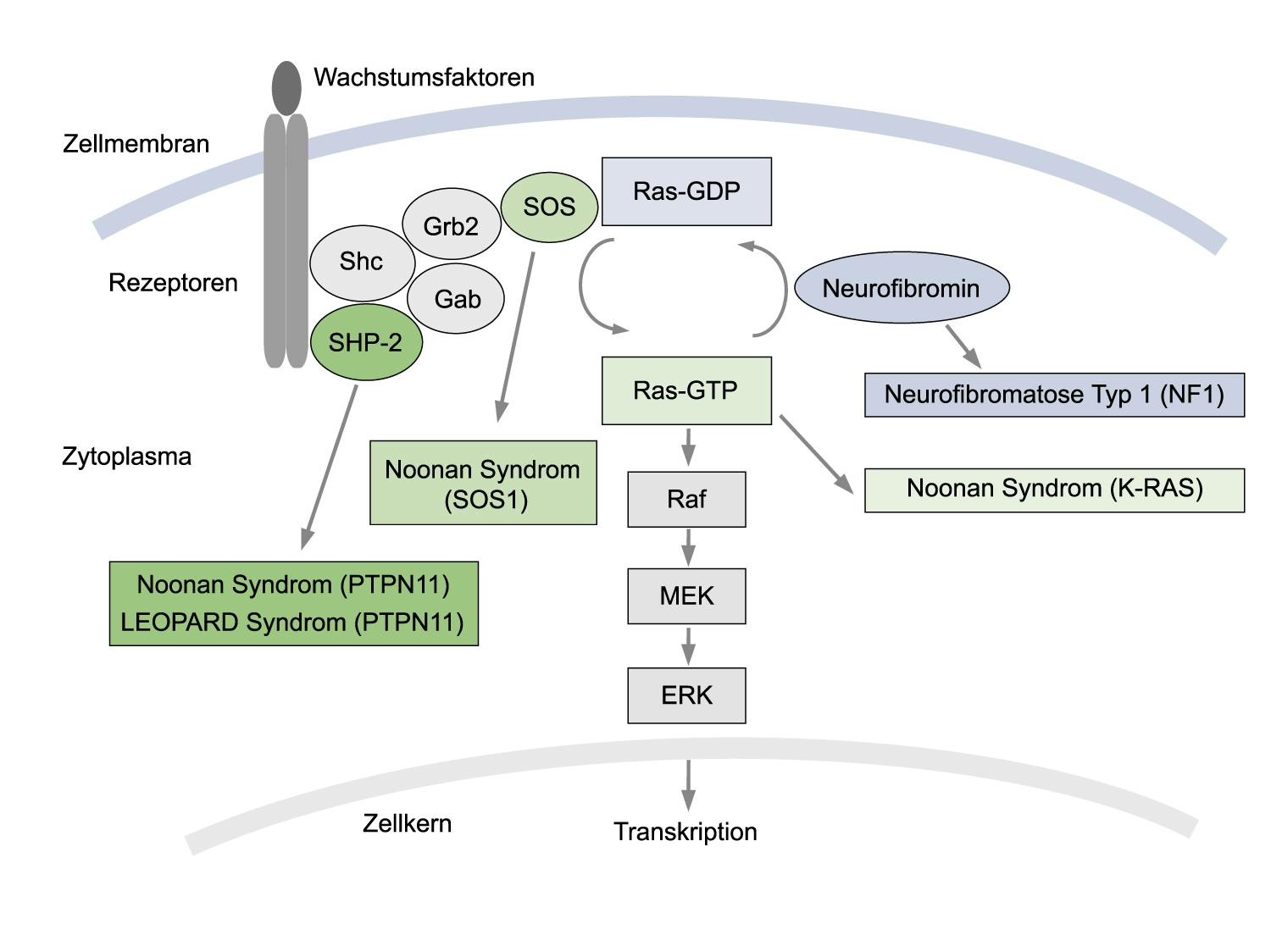
Bisher wurden pathogene Varianten in einigen weiteren Genen u.a. der RAS-ERK-MAP-Kinase-Signaltransduktion bei Noonan-Syndrom identifiziert. In bis zu 15% der Noonan-Patienten, die keine Mutation im PTPN11-Gen aufwiesen, wurden Mutationen im SOS1 (Son of Sevenless)-Gen nachgewiesen. Bei ca. 8% der Noonan-Patienten konnten pathogene Varianten im RAF1-Gen identifiziert werden, wobei fast alle Patienten mit RAF1-Mutationen eine Hypertrophe Kardiomyopathie aufweisen. Weitere Gene sind u.a. RIT1 (ca. 5% der Patienten), KRAS(ca. 3% der Patienten), wobei pathogene Varianten hier überwiegend zu schwerwiegenden Phänotypen führen.
Seltener werden bei Noonan-Syndrom pathogene Varianten in den Genen MAP2K1, MAP2K2 und BRAF identifiziert, wobei diese Gene häufiger bei Patienten mit Cardio-Facio-Cutanem (CFC) Syndrom verändert sind. Die Häufigkeit von pathogenen BRAF-Varianten bei Noonan-Syndrom wird auf 2% geschätzt. Hier weisen die beschriebenen Patienten neonatale Wachstumsverzögerung, leichte kognitive Defizite und Muskelhypotonie auf. Noch seltener sind mit 1% Varianten im NRAS- und im CBL-Gen. Weitere, im Zusammenhang mit Noonan-Syndrom beschriebene Gene sind u.a. RASA2, PPP1CB (Noonan-Syndrom-ähnliche Erkrankung mit losem Anagenhaar) und MRAS.
Varianten dieser o.g. Gene und der Gene MAP2K2, SHOC2, HRAS und NF1 finden sich u.a. auch bei Patienten mit Noonan-Syndrom-ähnlichen Erkrankungen, wie z.B. Cardio-Facio-Cutanes (CFC)-, LEOPARD-, Noonan-Syndrom mit juveniler myelomonozytärer Leukämie (JMML), Neurofibromatose-Noonan-Syndrom u.a. Pathogene Varianten im NF1-Gen wurden auch bei Noonan-Syndrom-Patienten, die die klinischen Kriterien einer klassischen Neurofibromatose (NF) nicht erfüllen, nachgewiesen.
Bei etwa 25% aller Patienten mit Noonan-Syndrom kann in den bisher bekannten Genen keine ursächliche Variante nachgewiesen werden.
Motta et al. 2019 Hum Mol Genet, ddz108, doi.org/10.1093/hmg/ddz108 / Bouchikhi et al. 2016 Int J of Ped and Adol Med 3:133 / Chen et al. 2014, PNAS 111:11473 / Vissers et al. 2014, Eur J Hum Genet doi:10.1038/ejhg.2014.115 / Aoki et al. 2013, Am J Hum Genet, 93:173-180 / Tartaglia et al. 2011, Best Pract Res Clin Endocrinol Metab 25:161 / Tartaglia et al. 2010, Mol Syndromol 1:2 / Cirstea et al. 2010, Nat Genet 42:27 / Sarkozy et al. 2009, Hum Mutat 30:695 / Nava et al. 2007, J Med Genet 44:763 / Razzaque et al. 2007, Nat Genet 39:1013 / Roberts et al. 2007, Nat Genet 39 / Carta et al. 2006, Am J Hum Genet 79: 129 / Schubbert et al. 2006, Nature Genet 38: 331 / Noonan 1968, Am J Dis Child 116:373
letzte Aktualisierung: 9.4.2024