Hypertrophe Kardiomyopathie (HCM)
Die hypertrophe Kardiomyopathie (HCM) ist eine autosomal-dominant vererbte Herzkrankheit, die durch eine asymmetrische Verdickung des linken Ventrikels gekennzeichnet ist und bei etwa 1 von 500 Menschen in der kaukasischen Bevölkerung vorkommt. Sie kann zu einem hohen Risiko für plötzlichen Herztod führen, wobei die Lebenserwartung im Durchschnitt 66 Jahre beträgt. Über 2.000 pathogene Varianten in mehr als 40 Genen, die hauptsächlich für kardiale Strukturproteine codieren, wurden im Zusammenhang mit HCM identifiziert, wobei etwa 90% dieser Varianten in den Genen MYH7, MYBPC3, TNNT2 und TNNI3 zu finden sind.
Wissenschaftlicher Hintergrund
Bei der hypertrophen Kardiomyopathie (HCM) handelt es sich um eine autosomal-dominant vererbte, strukturelle Erkrankung des Herzmuskels, die mit einer Prävalenz von ca. 1:500 in der kaukasischen Bevölkerung auftritt. In der Regel ist die hypertrophe Kardiomyopathie mit einer asymmetrisch erhöhten Muskelmasse des linken Ventrikels unter Beteiligung des interventrikulären Septums assoziiert, wodurch es zu charakteristischen Veränderungen im EKG kommt (Q-Welle, ST-Strecke und P-Welle).
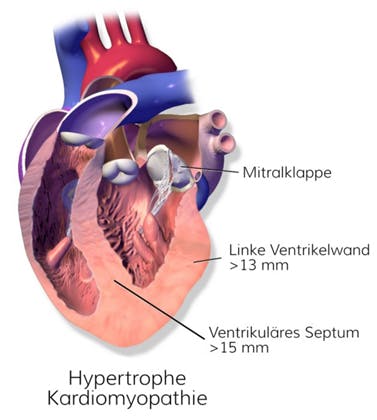
Abb.: Darstellung der strukturellen Veränderungen bei einem Herz mit hypertropher Kardiomyopathie (mod. nach By BruceBlaus (own work) [CC BY-SA 4.0], via Wikimedia Commons)
Die phänotypische Ausprägung der hypertrophen Kardiomyopathie variiert von benignen, unvollständig penetranten bis zu malignen Formen mit einem hohen Risiko für plötzlichen Herztod bereits im Kindesalter. Die durchschnittliche Lebenserwartung der Betroffenen liegt bei 66 Jahren, wobei die Prognose abhängig von der zugrunde liegenden molekularen Ursache ist.
Bislang wurden im Zusammenhang mit HCM ca. 2.000 pathogene Varianten in über 40 verschiedenen Genen, die meist für kardiale Struktur-Proteine codieren, identifiziert. Ca. 90% der bisher beschriebenen Varianten befinden sich in den Genen für die schwere Kette des ß-Myosins (MYH7), das Myosinbindeprotein-C (MYBPC3), Troponin T (TNNT2) und Troponin I (TNNI3). Insgesamt können derzeit im Rahmen der Routinediagnostik ursächliche Varianten in ca. 60% aller HCM-Fälle nachgewiesen werden. Deletionen einzelner Exons oder gesamter Gene sind sehr selten (<1% aller Fälle) und werden hier ebenfalls untersucht.
Zur Anforderung der erweiterten Diagnostik* bitte den Untersuchungsauftrag Herzerkrankungen verwenden.
*erweitert um Gene mit derzeit noch nicht hinreichend geklärter Krankheitsrelevanz (Gene unklarer Signifikanz).
Mazzarotto et al, J Am Heart Assoc e015473 (2020) / Walsh et al, Genet Med 19:192 (2017) / Elliott et al, Eur Heart J 35:2733 (2014) / Lopes et al, J Med Genet 50:228 (2013) / Ackerman et al, Europace 13:1077 (2011) / Rupp et al. 2018, Clin Res Cardiol doi.org/10.1007/s00392-018-1354-8 / Walsh et al. 2017, Genet Med 19:19
letzte Aktualisierung: 7.12.2023