Sphärozytose, hereditäre (Elliptozytose / Pyropoikilozytose inbegriffen)
Die hereditäre Sphärozytose ist eine häufige kongenitale hämolytische Anämie, die durch Defekte in Erythrozytenmembranproteinen verursacht wird. Diese führen zu Formverlust der Erythrozyten und erhöhter Membranpermeabilität. Symptome variieren von mild bis schwer und können Anämie, Ikterus und Splenomegalie umfassen. Die Krankheit ist genetisch heterogen, wobei die meisten Fälle autosomal-dominant vererbt werden und pathogene Varianten im ANK1-, SLC4A1-, SPTB-, SPTA1- und EPB42-Gen sein.
Wissenschaftlicher Hintergrund
Die hereditäre Sphärozytose (HS) ist die häufigste Ursache einer kongenitalen hämolytischen Anämie bei Kaukasiern. Die Prävalenz liegt bei ca. 1:2.000 bis 1:5.000. Die molekulare Ursache liegt in einem Defekt der Erythrozytenmembranproteine, die bei der Stabilisierung und Organisation der Plasmamembran eine zentrale Rolle spielen.
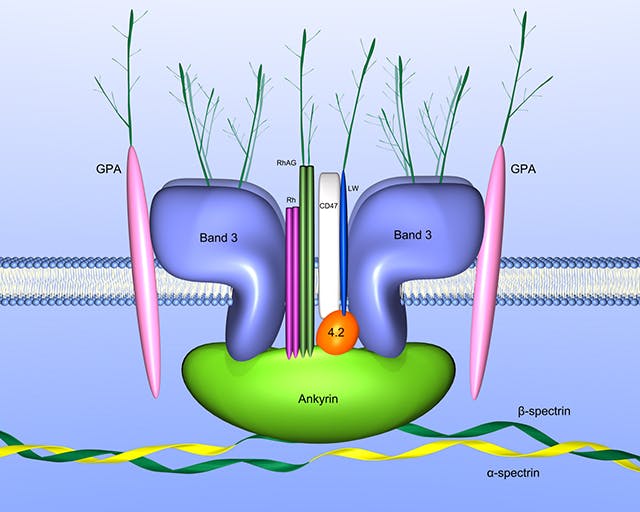
Ein Defekt in diesem Netzwerk führt zu einem Formverlust der Erythrozyten (Sphärozyten, Kugelzellen) und zu einer erhöhten Membranpermeabilität, gesteigerter Glykolyse und erhöhtem ATP-Umsatz. Die Sphärozyten werden frühzeitig über die Milz wieder aus dem Blutkreislauf entfernt.
Bei der hereditären Sphärozytose handelt es sich um ein klinisch, biochemisch und genetisch heterogenes Krankheitsbild. Betroffene zeigen hauptsächlich eine normozytäre hämolytische Anämie, Ikterus und Splenomegalie. Häufig finden sich auch Gallensteine. Die Klinik kann stark variieren von einer asymptomatischen Form bis hin zu einer schweren lebensbedrohlichen Anämie, die durch Transfusionen und Splenektomie behandelt werden muss.
Schweregrad | Anteil an Patienten (%) | Hämoglobin (g/dl) | Retikulozyten (%) | Bilirubin (mg/dl) | Sphärozyten (Blutausstrich) | Transfusionen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Leichte HS | 25–33 | 11,0–15,0 | 2,2–6 | 1–2 | oft nur vereinzelt | 0–1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mittelschwere HS | 60–70 | 8,0–11,0 | ≥ 6 | ≥ 2 | deutlich vermehrt | 0–2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schwere HS2 | ≈ 10 | 6,0–8,0 | ≥ 10 (meist > 15) | 2–3 | deutlich vermehrt | ≥ 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die Vererbung kann sowohl autosomal-dominant als auch rezessiv erfolgen, wobei ca. 2/3 aller Fälle einen dominanten Erbgang aufweisen. In Nordeuropa findet man bei bis zu 65% der Patienten pathogene Varianten im ANK1-Gen, welches für das Protein Ankyrin-1 codiert. Frameshift- bzw. Stopp-Mutationen führen dabei in der Regel zu einem dominanten Phänotyp und Missense- und Promotormutationen zu einem rezessiven Phänotyp. Als zweithäufigste Ursache sind ursächliche Varianten im SLC4A1-Gen beschrieben, die zu einem Defekt des Protein-Band-3 führen. Die Vererbung erfolgt autosomal-dominant. Des Weiteren sind in ca. 15-30% Varianten im β-Spectrin beschrieben, welches durch das SPTB-Gen codiert wird. In selteneren Fällen sind das α-Spectrin (SPTA1) oder das Protein 4.2 (EPB42) betroffen mit einem Anteil von jeweils unter 5%.
Sphärozytose Typ | Protein, Gen | Patienten mit HS | Erbgang | Schweregrad | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sphärozytose Typ 1 | Ankyrin-1, ANK1 | USA und Europa 40-65%; Japan 5-10% | AD, AR, de novo | mild bis moderat | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sphärozytose Typ 2 | β-Spectrin, SPTB | 15-30% | AD, de novo | mild bis moderat | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sphärozytose Typ 3 | α-Spectrin, SPTA1 | <5% | AR | schwer | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die Diagnostik der Sphärozytose basiert auf einer Kombination aus klinischen und laborchemischen Parametern.
Parameter (obligate Bestimmung) | Spezifizierung | Notwendigkeit der Untersuchung oder Bewertung des Nachweises (als diagnostisches Kriterium) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Familienanamnese | autosomal-dominant oder -rezessiv | fakultativ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Splenomegalie | körperliche Untersuchung Sonographie | fakultativ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Blutbild (maschinell) |
| fakultativ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die Untersuchung des SPTA1-, SPTB– und EPB41*-Gens kann auch bei V.a. auf eine hereditäre Elliptozytose oder Pyropoikilozytose durchgeführt werden, da diese ebenfalls durch Varianten in den genannten Genen bedingt werden.
Elliptozytose Typ | Gen | Protein | Erbgang | Anteil | Schweregrad | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elliptozytose Typ 1 (EL1) | EPB41* | Protein 4.1 | AD | 5% | asymptomatisch/mild bis moderat | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elliptozytose Typ 2 (EL2) | SPTA1 | α-Spectrin | AD | 65% | asymptomatisch/mild bis moderat | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elliptozytose Typ 3 (EL3) | SPTB | β-Spectrin | AD | 30% | asymptomatisch/mild bis moderat | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Zamora EA, Schaefer CA. Hereditary Spherocytosis. [Updated 2019 Mar 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing / Eber und Andres AWMF Leitlinie “Hereditäre Sphärozytose”, 2016 / Kalfa TA, Connor JA, Begtrup AH. EPB42-Related Hereditary Spherocytosis. 2014 Mar 13 [Updated 2016 Nov 10]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
letzte Aktualisierung: 12.11.2023