Ehlers-Danlos-Syndrom (EDS)
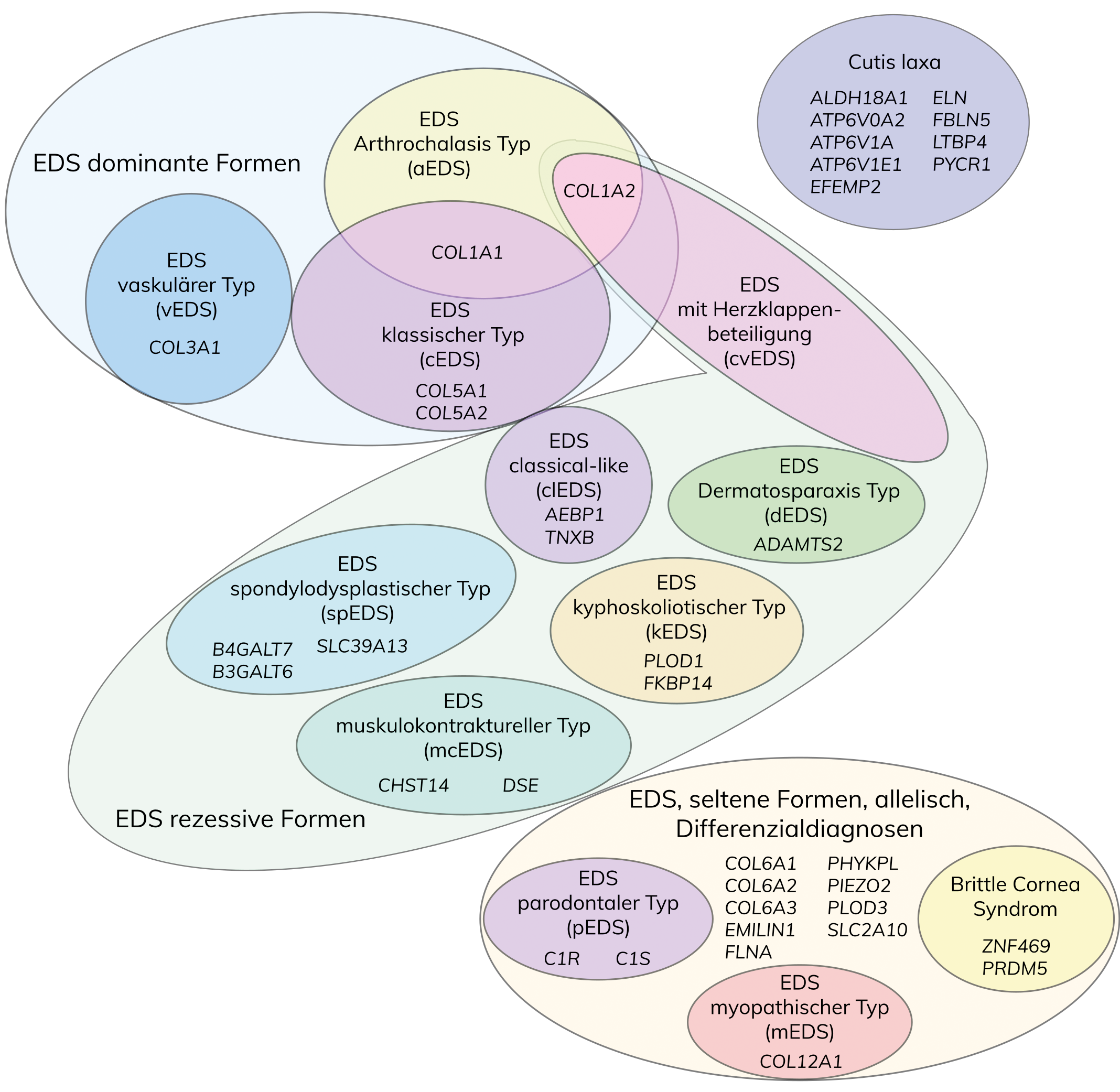
Das Ehlers-Danlos-Syndrom (EDS) ist eine Gruppe genetischer Erkrankungen des Bindegewebes, gekennzeichnet durch überbewegliche Gelenke, überdehnbare Haut und verletzliches Gewebe. Ursprünglich in sechs Haupttypen unterteilt, schlägt die revidierte Klassifikation von 2017 aufgrund neuer genetischer Erkenntnisse 13 Subtypen vor. Diese EDS-Subtypen basieren auf verschiedenen Defekten, insbesondere in Bezug auf Kollagen-Biosynthese, -Faltung und andere zelluläre Prozesse.
Wissenschaftlicher Hintergrund
Unter Ehlers-Danlos-Syndrom (EDS) wird eine klinisch und genetisch heterogene Gruppe von Erkrankungen des Bindegewebes zusammengefasst, die durch eine Überbeweglichkeit der Gelenke, überdehnbare Haut und verletzliches Gewebe charakterisiert ist.
Nach der vereinfachten Villefranche-Klassifikation von 1998 wurde EDS anhand klinischer, biochemischer und genetischer Daten in sechs Haupttypen unterteilt, die auf verschiedene molekulare Defekte des Kollagenstoffwechsels zurückzuführen sind. Infolge der Zunahme von genetisch aufgeklärten, neuen EDS-Subtypen schlägt die revidierte internationale Klassifikation von 2017 anhand klinischer Kriterien 13 Subtypen vor, die autosomal-dominant oder rezessiv vererbt werden und mit Ausnahme des EDS hypermobiler Typ durch genetische Daten bestätigt werden können. Anhand genetischer Ursachen und pathogenetischer Mechanismen können die einzelnen EDS-Subtypen auf der Grundlage von Defekten der Kollagen-Biosynthese und -Prozessierung, der Kollagen-Faltung und Prozessierung, Defekten der Struktur und Funktion der Verbindung zwischen Muskel und der extrazellulären Matrix, Defekten der Glycosaminoglycan-Biosynthese, des Komplement-Systems und intrazellulärer Prozesse in verschiedene Gruppen unterteilt werden.
Der sehr seltene Arthrochalasis EDS-Subtyp (aEDS) wird autosomal-dominant vererbt. Charakteristisch sind eine extreme, generalisierte Überstreckbarkeit der Gelenke, verbunden mit Gelenksubluxationen sowie eine angeborene, beidseitige Hüftluxation. Zusätzliche Symptome können Muskelhypotonie, Kleinwuchs, Osteopenie, Kyphoskoliose und samtige, hyperelastische Haut sein. Im Gegensatz zu anderen EDS-Subtypen können milde Dysmorphiezeichen wie Hypertelorismus, bilaterale Epikanthus-Falten, breite Fontanellen und Mikrognathie auftreten.
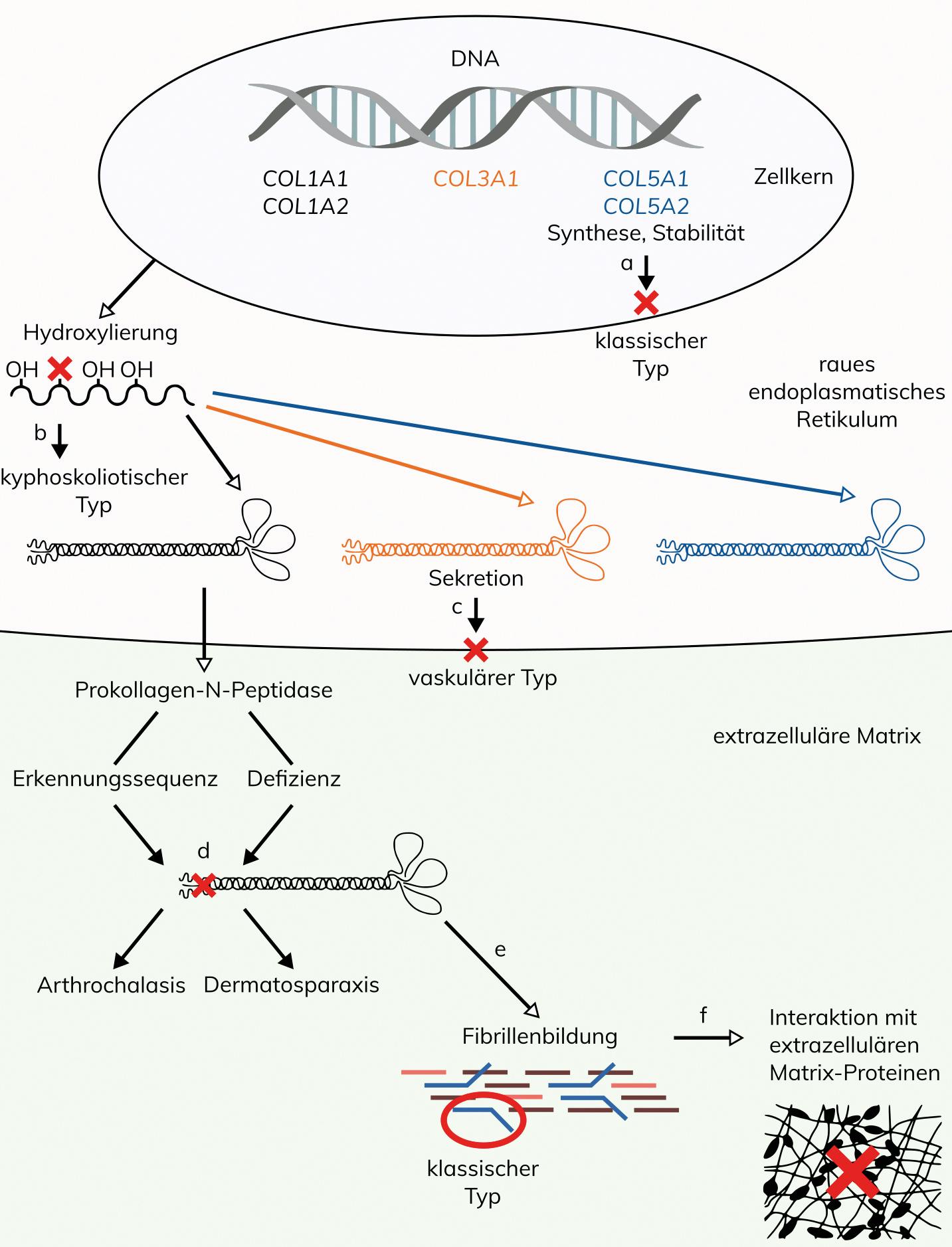
Ursache sind Varianten in den Kollagen-Genen COL1A1 und COL1A2, die für die α1- und α2-Ketten des in der Haut und im Knochen vorherrschenden Typ I-Kollagens codieren. Fast alle der beschriebenen Varianten betreffen jeweils Exon 6 des COL1A1-Gens bzw. des COL1A2-Gens. Dies sind vor allem Spleißvarianten, wodurch die Erkennungsstelle für die Abspaltung der N-terminalen Propeptide in den α-Ketten des Typ I-Prokollagens eliminiert wird. Daneben ist jeweils eine genomische Deletion im COL1A1-Gen bzw. im COL1A2-Gen beschrieben, die Exon 6 einschließt, und ebenfalls wie die Spleißvarianten die Erkennungsstelle für die Abspaltung der N-terminalen Propeptide eliminiert. Bisher ist außerhalb dieser definierten Regionen bei Patienten mit aEDS nur eine partielle Duplikation des COL1A2-Gens beschrieben, wobei der betroffene Patient zusätzlich Symptome einer Osteogenesis imperfecta aufwies. Insgesamt sind bei aEDS vier verschiedene pathogene Varianten im COL1A1-Gen und 12 verschiede pathogene Varianten im COL1A2-Gen beschrieben. Unvollständig prozessierte Prokollagen-Ketten können auch biochemisch und elektronenmikroskopisch durch eine charakteristische Ultrastruktur nachgewiesen werden.
In der aktuellen EDS-Klassifikation von 2017 wird der autosomal-rezessiv vererbte EDS-Typ mit Tenascin-X-Defizienz als dem klassischen Typ ähnliches (classical like) Ehlers-Danlos-Syndrom (clEDS) bezeichnet. Die Minimalanforderung für die klinische Diagnosestellung clEDS ist das Vorliegen aller drei Hauptkriterien: überdehnbare, samtige Haut ohne atrophe Narbenbildung, generalisierte Hypermobilität der Gelenke ohne Dislokationen und erhöhte Verletzlichkeit der Haut mit spontanen Ecchymosen. Somit bestehen bzgl. der Gelenk- und Hautbeteiligung klinische Überlappungen zum klassischen EDS (cEDS), allerdings ohne Zigarettenpapier-artige Narbenbildung, und zum hypermobilen EDS (hEDS), der wiederum keine erhöhte Verletzlichkeit der Haut aufweist.
Ehlers-Danlos-Syndrom, classic like Typ 1 (EDSCLL)
EDS mit Tenascin-X-Defizienz wurde anhand eines Patienten mit Adrenogenitalem Syndrom und einer 21-Hydroxylase-Defizienz genetisch aufgeklärt, der zusätzlich klinische Symptome eines klassischen EDS aufwies. Die Ursache war eine 30-kb-Deletion auf Chromosom 6p21.3, die sowohl das CYP21A2-Gen als auch das teilweise überlappende TNXB-Gen beinhaltete und somit ein Contiguous Gene Syndrome darstellte. Molekulare Ursache einer Tenascin-X-Defizienz sind homozygote oder kombiniert heterozygote Varianten im TNXB–Gen. Bisher sind in der Human Gene Mutation Database und der Ehlers-Danlos Syndrome Variant Database (LOVD) im TNXB-Gen erst sechs pathogene Missense-Varianten, sechs Nonsense-Varianten, fünf kleine Frameshift-Varianten, zwei Spleißvarianten sowie zwei große genomische Rearrangements enthalten. Homozygotie und kombinierte Heterozygotie von Varianten, die zum vorzeitigen translationalen Stop führen, resultieren in einem kompletten Fehlen des Genprodukts Tenascin-X auf RNA- und Proteinebene. Bei den Patienten ist kein Tenascin-X im Serum mehr nachweisbar. Tenascin-X ist ein Glykoprotein, das in der extrazellulären Matrix von Haut, Sehnen, Muskeln und Blutgefäßen synthetisiert wird. Einige Patienten mit Tenascin-X-Defizienz weisen myopathische Symptome auf, die charakteristisch für eine Bethlem Myopathie oder eine Ullrich-Muskeldystrophie sind. Das Fehlen von Tenascin-X im Serum bewirkt eine verminderte Expression von Typ VI-Kollagen.
Der fehlende Nachweis von Tenascin-X im Serum unterstützt die klinische Diagnose EDS mit Tenascin-X-Defizienz.
Ehlers-Danlos-Syndrom, classic like Typ 2 (EDSCLL2)
2018 wurde eine weitere, autosomal-rezessiv-vererbte Form des classic like Subtyps (EDSCLL2) beschrieben. Charakteristisch ist eine massive Haut- und muskuloskelettale Beteiligung mit phänotypischer Variabilität und klinischer Überlappung zu anderen EDS-Subtypen. Die Hautmanifestationen beinhalten hyperelastische, schlaffe, verletzliche Haut, die zum Teil auch durchscheinend sein kann, mit verzögerter Wundheilung und atropher Narbenbildung. Neben einer generalisierten Gelenkhypermobilität, Gelenkdislokationen und Subluxationen ist eine frühmanifestierende Osteoporose oder Osteopenie charakteristisch. Zusätzlich sind kardiovaskuläre Komplikationen beschrieben. Die Ultrastruktur einer Hautbiopsie bei den bisher untersuchten Patienten wies unregelmäßige Querschnitte der Kollagenfibrillen und ausgefranste Kollagenfasern auf.
Molekulare Ursache sind homozygote oder kombiniert heterozygote loss of function Varianten im AEPB1-Gen. AEBP1 codiert für das aortale Carboxypeptidase-ähnliche Protein ACLP, welches innerhalb der extrazellulären Matrix (ECM) mit Kollagen assoziiert ist und an der Proliferation von Fibroblasten und mesenchymalen Stammzellen zu Kollagen-produzierenden Zellen beteiligt ist. Eine Hypothese zur Pathophysiologie einer ACLP-Defizienz ist eine abnorme Zusammenlagerung der Kollagenfibrillen und eine gestörte Wundheilung durch eine reduzierte TGFβ-Rezeptor-Signalübertragung.
Das sehr seltene, autosomal-rezessiv vererbte EDS Dermatosparaxis Typ (dEDS) ist durch eine extrem verletzliche, schlaffe Haut charakterisiert, die besonders im Gesicht überschüssig erscheint und an Cutis laxa erinnert. Weitere Symptome sind Hämatomneigung, vorzeitige Ruptur fetaler Membranen, Fragilität innerer Organe, große Nabel- und Leistenhernien, sowie Kleinwuchs und kurze Finger.
Die molekulare Ursache ist eine Prokollagen I-N-Proteinase Defizienz, die bei der Reifung der pro-α1 (I) und pro-α2 (I) Kollagenketten zum Einbau der unreifen pNa1(I) und pNa2(I) Pro-Kollagenketten in die Kollagenfibrillen führt. Der Aufbau der Kollagenfibrillen wird dadurch derart gestört, dass im Querschnitt der Dermis in der Elektronenmikroskopie pathognomonische Hieroglyphen-artige Strukturen erkennbar sind.
Das ADAMTS2-Gen codiert für die Prokollagen I-N-Proteinase, eine Zink-Metalloproteinase der ADAMTS-Familie, welche die Aminopropeptide der Typ I-, Typ II- und Typ III-Prokollagene abspalten. ADAMTS (A Disintegrin-like And Metalloproteinase with ThromboSpondin type 1 motif) sind Anker-Proteine der extrazellulären Matrix (EZM). Bisher sind erst 11 verschiedene, inaktivierende ADAMTS2-Varianten beschrieben, die zum vorzeitigen translationalen Stop führen oder genomische Rearrangements beinhalten. Die Varianten liegen meistens homozygot und seltener kombiniert heterozygot vor.
EDS klassischer Typ (cEDS) wird autosomal-dominant vererbt. Mit einer Prävalenz von 1:20.000 ist er der zweithäufigste EDS-Subtyp. In den Erstbeschreibungen wurde zwischen EDS gravis und EDS mitis differenziert, die sich nur durch den Schweregrad unterscheiden. Gemäß der Klassifikation von 2017 sind eine signifikante Überdehnbarkeit der Haut mit einer atrophen Narbenbildung (Zigarettenpapiernarben) und eine generalisierte Überbeweglichkeit der Gelenke die klinischen Hauptkriterien. Nebenkriterien sind Hämatomneigung, weiche, samtige Haut, Gewebeverletzlichkeit, molluscoide Pseudotumore, subkutane Spheroide, Hernien, Epikantusfalten sowie Komplikationen des Bewegungsapparats durch die Überbeweglichkeit und Komplikationen bei Operationen aufgrund der Gewebebrüchigkeit.
Die molekulare Ursache bei Patienten mit Ehlers-Danlos-Syndrom klassischer Typ (cEDS) sind Varianten in den Genen COL5A1 und COL5A2, welche für die α1- und α2-Kette des Typ V-Kollagens codieren. Bei etwa 75% der Patienten liegen Varianten im COL5A1-Gen vor, bei etwa 14% der Patienten wurden Varianten im COL5A2-Gen beschrieben. Gemäß der Klassifikation von 2017 ist die Minimalanforderung für eine genetische Diagnostik bei V.a. cEDS als klinisches Hauptkriterium eine signifikante Überdehnbarkeit der Haut mit einer atrophen Narbenbildung, die entweder in Kombination mit einer generalisierten Überbeweglichkeit der Gelenke als weiteres klinisches Hauptkriterien oder mit mindestens drei klinischen Nebenkriterien vorliegt. Bei Patienten, bei denen alle Hauptkriterien erfüllt waren, konnten in 90% pathogene Varianten in den Genen COL5A1 und COL5A2 nachgewiesen werden. Der Großteil aller COL5A1– und COL5A2-Varianten führen zum vorzeitigen translationalen Stop und in der Folge zu einem Null-Allel. Etwa 30% aller COL5A1-Varianten und 40% aller COL5A2-Varianten sind struktureller Art, wobei Glycin in der Tripel-Helix betroffen ist. Genomische Deletionen wurden im COL5A2-Gen bisher nicht identifiziert, im COL5A1-Gen sind in der Literatur erst vier genomische Deletionen und eine genomische Duplikation beschrieben.
Die klinische Diagnose cEDS kann durch den Nachweis einer pathogenen COL5A1– oder COL5A2-Variante gesichert werden. Bei fehlendem molekulargenetischen Nachweis kann eine charakteristische Ultrastruktur mit blumenkohlartigen Veränderungen in der elektronenmikroskopischen Untersuchung der Kollagenfibrillen einer Hautbiopsie die klinische Diagnose cEDS unterstützen.
Bei etwa 1% der Patienten mit cEDS wurden auch Varianten im COL1A1-Gen identifiziert. Sie betreffen entweder Arginin-Substitutionen und sind dann häufig mit Gefäßbeteiligung assoziiert, während EDS-Patienten mit Glycin-Substitutionen, anderen Aminosäureaustauschen oder translationalen Stopmutationen im COL1A1-Gen zusätzlich Symptome einer Osteogenesis imperfecta aufweisen.
Der autosomal-rezessiv vererbte, kyphoskoliotische EDS-Typ (kEDS) ist genetisch heterogen. Charakteristische klinische Symptome sind Kyphoskoliose, Muskelhypotonie, überdehnbare dünne verletzliche Haut, atrophe Narbenbildung, überbewegliche Gelenke und variable Augenbeteiligung. Weiterhin verschiedene kraniofaziale Auffälligkeiten, Gelenkkontrakturen und runzelige Handflächen.
Bei einem Großteil der Patienten wird die Erkrankung durch homozygote oder kombiniert heterozygote Varianten im PLOD1-Gen verursacht, das für das Enzym Lysylhydroxylase 1 (LH) codiert (kEDS-PLOD1). LH ist für die Hydroxylysin-abhängige Pyridinolin-Quervernetzung von Typ I und Typ III Kollagen verantwortlich, das hauptsächlich im Skelett zu finden ist. Das Fehlen des Enzyms LH kann auch durch ein erhöhtes Verhältnis der Quervernetzungen von Lysyl-Pyridinolin (LP) zu Hydroxylysyl-Pyrodinolin (HP) im Urin nachgewiesen werden.
Ein kleiner Teil der Patienten mit nahezu identischer Klinik hat einen unauffälligen LP/HP-Quotienten im Urin und keine PLOD1-Varianten. 2012 wurde differenzialdiagnostisch zu kEDS-PLOD1 eine weitere autosomal-rezessive EDS-Form mit unauffälligem LP/HP-Quotienten beschrieben und zunächst als EDSKMH bezeichnet. Hier ist neben progressiver Kyphoskoliose, Muskelhypotonie, Gelenküberbeweglichkeit, hyperelastischer Haut und Myopathie besonders der sensineurale Hörverlust charakteristisch. Infolge der klinischen Überlappung mit kEDS-PLOD1 wird EDSKMH in der aktuellen EDS-Klassifikation ebenfalls in die Gruppe EDS kyphoskoliotischer Typ (kEDS) eingeordnet und als kEDS-FKPB14 bezeichnet. Ursache sind Varianten im FKBP14–Gen, das für das FK506-binding protein 22, einem Mitglied der Peptidyl-Prolyl cis-trans Isomerasen (PPIasen) codiert.
Der sehr seltene, autosomal-rezessiv vererbte EDS-Typ mit Herzklappenbeteiligung (cvEDS) ist durch eine schwerwiegende Herzklappenbeteiligung mit Mitralklappeninsuffizienz, Aortenklappeninsuffizienz, Vorhofseptumdefekt, Ventrikelerweiterung und Ventrikelhypertrophie gekennzeichnet, die in der Regel einen Mitral- und Aortenklappenersatz notwendig machen. Weitere EDS-spezifische Symptome sind eine variable Überdehnbarkeit der Haut, atrophe Narbenbildung und eine generalisierte Gelenküberbeweglichkeit.
Bisher sind erst acht Patienten aus sechs Familien beschrieben, bei denen homozygote oder kombiniert heterozygote Varianten im COL1A2-Gen identifiziert wurden. Dabei handelt es sich um sog. Nullallele, die zu einer Instabilität der Transkripte führen, so dass in einer Kollagenelektrophorese keine Pro-α2(I)-Kollagenketten mehr nachweisbar sind. Im Gegensatz zu Patienten mit Osteogenesis imperfecta liegt bei cvEDS keine erhöhte Knochenbrüchigkeit vor.
Der autosomal-rezessiv vererbte, muskulokontrakturelle EDS-Typ (mcEDS) ist genetisch heterogen und wurde in der historischen Villfranche-Klassifikation zu der Gruppe des kyphoskoliotischen EDS-Typs mit unauffälligem LP/HP-Quotienten (EDS Typ VIB) gezählt. Mittlerweile unterscheidet man bei mcEDS Patienten mit einer D4ST1-Defizienz und Patienten mit einer DSE-Defizienz.
mcEDS ist eine Differenzialdiagnose zu neuromuskulären Erkrankungen innerhalb des Spektrums von Bindegewebserkrankungen. Charakteristisch ist ein asthenischer Körperbau, eine Instabilität der großen Gelenke, verjüngende Fingerendglieder, eine Brachyzephalie mit charakteristischen Gesichtszügen, überdehnbare und verletzliche Haut, sowie rezidivierende subkutane Hämatome.
Molekulare Ursache einer D4ST1-Defizienz sind homozygote oder kombiniert heterozygote Varianten im CHST14-Gen (Carbohydrat Sulfotransferase 14), das für Dermatan 4-O-Sulfotransferase 1 (D4ST1) codiert. Molekulare Ursache einer DSE-Defizienz sind homozygote oder kombiniert heterozygote Varianten im DSE-Gen, das für die Dermatan-Sulfat-Epimerase (DSE) codiert. DSE katalysiert die Epimerisation von D-Glucuronsäure (D-GlcA) zu L-Iduronsäure (L-IdoA). Dadurch kann D4ST1 die 4-O-Sulfonierung des benachbarten N-Acetyl-D-Galactosamin katalysieren. Beide Enzyme sind an der Dermatan-Sulfat (DS)-Biosynthese beteiligt.
EDS myopathischer Typ (mEDS) ist gekennzeichnet durch eine Muskelschwäche, die sich in der Kindheit mit Kontrakturen der proximalen großen Gelenke und einer distalen Hypermobilität manifestiert. In der Regel verbessert sich die Muskelschwäche im jungen Erwachsenenalter, wobei es in der vierten Lebensdekade wieder zu einer Verschlechterung kommen kann. Das Spektrum der Erkrankungen, die durch Muskelschwäche, Hypotonie, Myopathie und eine Bindegewebssymptomatik charakterisiert sind, wurde ursprünglich mit Kollagen-Typ VI-Erkrankungen in Verbindung gebracht.
Molekulare Ursache für mEDS sind pathogene Varianten in COL12A1, das für die α1-Kette des Typ XII-Kollagens codiert. Kollagen XII liegt als Homotrimer auf der Oberfläche von Typ I-Kollagen-Fasern vor. Es bildet eine Verbindung zwischen Typ I-Kollagen-Fasern und Komponenten der extrazellulären Matrix wie Decorin, Fibromodulin und Tenascin. Bisher sind bei mEDS sowohl autosomal-dominant vererbte Formen mit heterozygoten Missense-Varianten als auch autosomal-rezessiv vererbte Formen mit homozygoten Frameshift-Varianten beschrieben.
Der autosomal dominant vererbte parodontale Typ des EDS (pEDS) ist gekennzeichnet durch eine schwere, früh einsetzende parodontale Entzündung. Diese beginnt in der Kindheit mit einer extensiven Gingivitis und führt im Jugendlichenalter zur Zerstörung des Zahnhalteapparats und zum vorzeitigen Zahnverlust. Weitere klinische Merkmale sind prätibiale Hyperpigmentierung, Akrogerie, verletzliche Haut und Zahnfleisch, veränderte Narbenbildung, generalisierte Überstreckbarkeit der Gelenke und eine Hämatomneigung. In Einzelfällen sind auch Rupturen von Arterien und inneren Organen sowie eine ZNS-Beteiligung in Form einer Mikroangiopathie und Leukenzephalopathie beschrieben.
2003 wurde eine Kandidatenregion auf Chromosom 12p13.1 lokalisiert, aber kein Gen identifiziert. 2016 konnten anhand von 19 Familien mittels ergänzender Exom-Analyse bei 15 Familien pathogene Varianten in C1Rund bei zwei Familien in C1Sidentifiziert werden. Die in der Region 12p13.1 direkt benachbarten Gene C1R und C1S codieren für die Untereinheiten von C1r und C1s des Komplementsystems. Die Proteine bilden ein Heterotetramer, das sich mit sechs C1q-Untereinheiten zusammenlagert. Die bisher identifizierten pathogenen Varianten führen zur intrazellulären Retention des Komplement-Komplexes und zu einer Vergrößerung des endoplasmatischen Retikulums.
EDS infolge B4GALT7-Defizienz (spEDS-B4GALT7) und B3GALT6-Defizienz (spEDS-B3GALT6)
Charakteristische klinische Symptome der bisher als progeroide Subtypen bezeichneten EDS-Syndrome sind ein vorgealtertes Aussehen, Entwicklungsverzögerung, Kleinwuchs, kraniofaziale Dysproportion, generalisierte Osteopenie, gestörte Wundheilung, hypermobile Gelenke, Muskelhypotonie und laxe, aber elastische Haut.
Molekulare Ursache sind homozygote oder kombiniert heterozygote Varianten in den Genen B4GALT7 und B3GALT6. Beide Gene codieren für eine UDP-Galactose:O-beta-D-Xylosylprotein 4-beta-D-Galactosyltransferase bzw. beta-1,3-Galactosyltransferase 6. Ein Galactosyltransferase-I-Mangel führt zu einer Defizienz von kleinen Proteodermatansulfaten (PDS) innerhalb der Glycosaminoglykan-Biosynthese. B3GALT6-Varianten wurden ursprünglich bei spondyloepimetaphysärer Dysplasie mit Überstreckbarkeit der Gelenke (SEMDJL1) identifiziert.
EDS infolge von SLC39A13-Varianten (spEDS-SLC39A13)
Patienten mit dem sehr seltenen, ehemals als Spondylocheirodysplastische Form (SCD-EDS) bezeichneten EDS-Typ weisen dünne, durchscheinende, hyperelastische, samtige, verletzliche Haut mit atrophen Narben, schlanke, spitz zulaufende Finger und Kontrakturen der Endgelenke auf. Charakteristische zusätzliche klinische Symptome sind eine Skelettdysplasie (spondylo) mit moderaten Kleinwuchs und charakteristische Handauffälligkeiten mit runzeligen Handflächen und Atrophie der Daumen- und Kleinfingerballen (cheiro).
spEDS-SLC39A13 wird durch Varianten im Zink-Transporter-Gen SLC39A13verursacht. Bisher sind erst drei homozygote los-of-function Varianten im SLC39A13-Gen bei neun Patienten aus drei Familien beschrieben. Bei Patienten mit spEDS-SLC39A13 findet sich im Urin ein Verhältnis von LP/HP von etwa 1.0, und damit ein Wert, der zwischen dem von Kontrollen und dem von Patienten mit dem kyphoskoliotischen EDS-Typ (kEDS-PLOD1) liegt.
Charakteristisch für den EDS vaskulärer Typ (vEDS) ist ein hohes Risiko für Rupturen von Arterien, Uterus und innerer Organe mit fatalen Blutungen. In den Wänden von Arterien und inneren Organen stellt Typ III-Kollagen mit bis zu 45% einen wichtigen Bestandteil dar, so dass diese Gewebe bei vEDS Typ besonders betroffen sind. Klinische Hauptkriterien gemäß der Klassifikation von 2017 sind eine Arterienruptur in jungem Alter, eine spontane Perforation des Colon sigmoideum, eine Uterusruptur im dritten Trimenon, AV-Fisteln zwischen A. carotis und Sinus cavernosus und eine positive Familienanamnese mit einer nachgewiesenen pathogenen COL3A1-Variante. Der Erbgang ist autosomal-dominant, die Prävalenz wird auf 1:50.000 geschätzt.
Bei Patienten mit dem vaskulären Ehlers-Danlos-Syndrom (vEDS) sind bisher ausschließlich pathogene Varianten im COL3A1-Gen veröffentlicht, die biochemisch zu einer veränderten Synthese, Struktur, oder Sekretion von Typ III-Prokollagen führen. Die Zusammensetzung der identifizierten Arten von Nukleotidveränderungen im COL3A1-Gen umfasst mit etwa 65% Glycin-Substitutionen innerhalb der Tripel-Helix-Domäne der pro-α1 (III)-Kette, mit 30% Varianten, die zum vorzeitigen translationalen Stop führen und die zu strukturell veränderten oder instabilen Polypeptiden führen, und mit weniger als 5% genomische Deletionen und komplexe Rearrangements, die ganze bzw. mehrere Exons betreffen. Dabei hat die Art der Variante einen Einfluss auf den klinischen Verlauf. So treten bei Patienten mit Null-Allelen vaskuläre Komplikationen später auf und die Lebenserwartung ist im Vergleich zu Patienten mit anderen Varianten erhöht, während Patienten mit Glycin-Substitutionen und Exon-Skipping-Varianten die ungünstigste Prognose haben. Die Lokalisation der Variante innerhalb des Gens hat dabei keinen Einfluss.
Indikationen für eine genetische Diagnostik bei V.a. vEDS sind eine positive Familienanamnese sowie das Vorliegen einer Arterien-Ruptur oder Dissektion vor dem 40. Lebensjahr, einer spontanen Sigmaperforation oder eines Spontanpneumothorax. Die klinische Diagnose vEDS kann durch den Nachweis einer pathogenen Variante im COL3A1-Gen oder einer veränderten Synthese, Struktur, oder Sekretion von Typ III-Prokollagen in kultivierten Hautfibroblasten gesichert werden. Obwohl COL3A1 das einzige bei vEDS veränderte Gen ist, bleibt die genetische Aufklärungsrate oft niedrig, weil der klinische Phänotyp bei den Patienten häufig unvollständig ist. Die Erfassungsrate im COL3A1-Gen liegt in der Literatur bei biochemisch nachgewiesenem, strukturell verändertem Typ III-Kollagen bei 95-100%. Bei Patienten mit klinischer Diagnose vEDS ohne Nachweis einer pathogenen COL3A1-Variante kann die Analyse anderer Gene des TGFß-Signaltransduktionsweges in Erwägung gezogen werden.
Cortini et al. 2019, Arch Dermatol Res 311:265 / Syx et al. 2019, Hum Mol Genet 28:1853 / Ritelli et al. 2019, Genes (Basel) 10, pii:E135 / Giunta in: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® (May 23, 2019) / Blackburn et al. 2018, Am J Hum Genet 102:696 / Weerakkody et al. 2018, Genet Med 18:1119 / Giunta et al. 2018, Genet Med 20:42 / Van Damme et al. 2018, Hum Mol Genet 27:3475 / Yeowell and Steinmann in: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® (updated 2018 Oct 18) / Wu et al. 2018, J Clin Periodontol 45:1311 / Malfait et al. in: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews® (Updated 2018 Jul 26) / Mayer in Luttkus (Hrsg.) 2018: Das Ehlers-Danlos-Syndrom, Kap. 3, 2. Auflage, De Gruyter / Demirdas et al. 2017, Clin Genet 91:411 / Chen et al. 2016, Hum Mutat 37(9):893 / Van Damme et al. 2016, Genet Med 18: / Kapferer-Seebacher et al. 2016, Am J Hum Genet 99:1005 / Morissette et al. 2015, J Clin Endocrinol Metab 100:E1143 / Frank et al. 2015, Eur J Hum Genet 23:1657 / Pepin et al. 2014, Genet Med 16:881 / Vorster et al. 2014, Clin Genet epub / Zou et al. 2014, Hum Mol Genet 23:2339 / Hicks et al. 2014, Hum Mol Genet 23:2353 / Guo et al. 2013, Am J Med Genet A 161:2519 / Nakajima et al. 2013, Am J Hum Genet 92:927 / Pénisson-Besnier et al. 2013, Neuromuscul Disord 23:664 / Merke et al. 2013, J Clin Endocrinol Metab 98:E379 / Reinstein et al. 2013, Eur J Hum Genet 21: 233 / Ritelli et al. 2013, Orphanet J Rare Dis 8:58 / Mayer in eLS 2012, John Wiley & Sons Ltd: Chichester [DOI: 10.1002/9780470015902.a0024295] / Klaassens et al. 2012, Clin Genet 82:121 / Symoens et al. 2012, Hum Mutat 33:1485 / Baumann et al. 2012, Am J Hum Genet 90:201 / Ong et al. 2012, Virchows Arch 460:637 / Rohrbach et al. 2011, Orphanet J Rare Dis 6:46 / Giunta et al 2008, Am J Hum Genet 82:1290 / Fukada et al. 2008, PLoS One 3:e3642 / Giunta et al. 2008, Am Med Genet 146A:1341 / Malfait et al. 2007, Genet Med 12:597 / Germain 2007, Orphanet J Rare Dis 2:32 / Giunta et al. 2005, Mol Genet Metab 86:269 / Colige et al. 2004, J Invest Dermatol 123:656 / Faiyaz-Ul-Haque et al. 2004, Am J Med Genet 128A:39 / Rahman et al. 2003, Am J Hum Genet 73: 198 / Nicholls et al. 2001, J Med Genet 38:132 / Schalkwijk et al. 2001, N Engl J Med 345:1167 / Yeowell et al. 2000, Mol Genet Metab 71:212 / Raff et al. 2000, Hum Genet 106:19 / Nicholls et al. 2000, J Med Genet 37:E33 / Pepin et al. 2000, N Engl J Med 342:673 / Okajima et al. 1999, J Biol Chem 274:28841 / Beighton et al. 1998, Am J Med Genet 77:31 / Byers et al. 1997, Am J Med Genet 72:94 / Hausser et al. 1994, Hum Genet 93:394 / Hartsfield et al. 1990, Am J Med Genet 37: 465 / Stewart et al. 1977, Birth Defects Orig Art Ser XIII:85
letzte Aktualisierung: 24.3.2024