




Qualifikation nach den Richtlinien der Gendiagnostik-Kommission (GEKO) – Aufgrund der Nachfrage bietet die Berlin-Lichtenberg GmbH ein weiteres Angebot zur Qualifikation “Fachgebunde Genetische Beratung” in 4 Modulen an.

Medicover Genetics hat erfolgreich die MVZ Humangenetik Köln GmbH übernommen und erweitert damit sein Netzwerk von genetischen Beratungszentren und Laboren in Deutschland.

Die CYP2C19-Genotypisierung ist ein wichtiger Baustein zur Dosisfindung bei der Mavacamten-Therapie und trägt somit zur Vermeidung von Nebenwirkungen bei.
Unser vielfältiges Portfolio spiegelt unser Engagement zur Qualität, Innovation und Patientenversorgung wider. Wir bieten außergewöhnlichen Service in der klinischen und genetischen Labordiagnostik an, um gezielte Diagnosen und personalisierte Therapieplanung zu ermöglichen.
Mit modernster Ausstattung und einem Team hochqualifizierter Fachleute bietet Medicover Diagnostics präzise und zeitnahe diagnostische Lösungen an. Wir führen ein breites Spektrum von Routine- bis hin zu fortgeschrittenen Tests durch, einschließlich Allergiediagnostik, Autoimmundiagnostik, Immunologie, Infektiologie/Mikrobiologie und Klinische Chemie, um Gesundheitsdienstleistern bei der bestmöglichen Versorgung ihrer Patient:innen zu unterstützen.
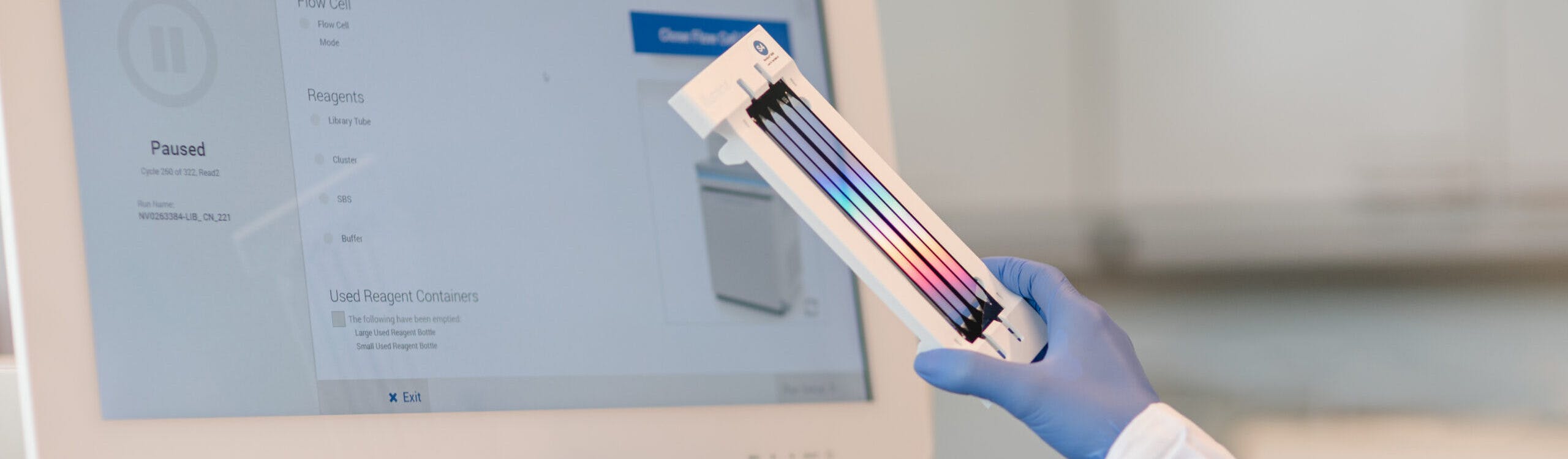
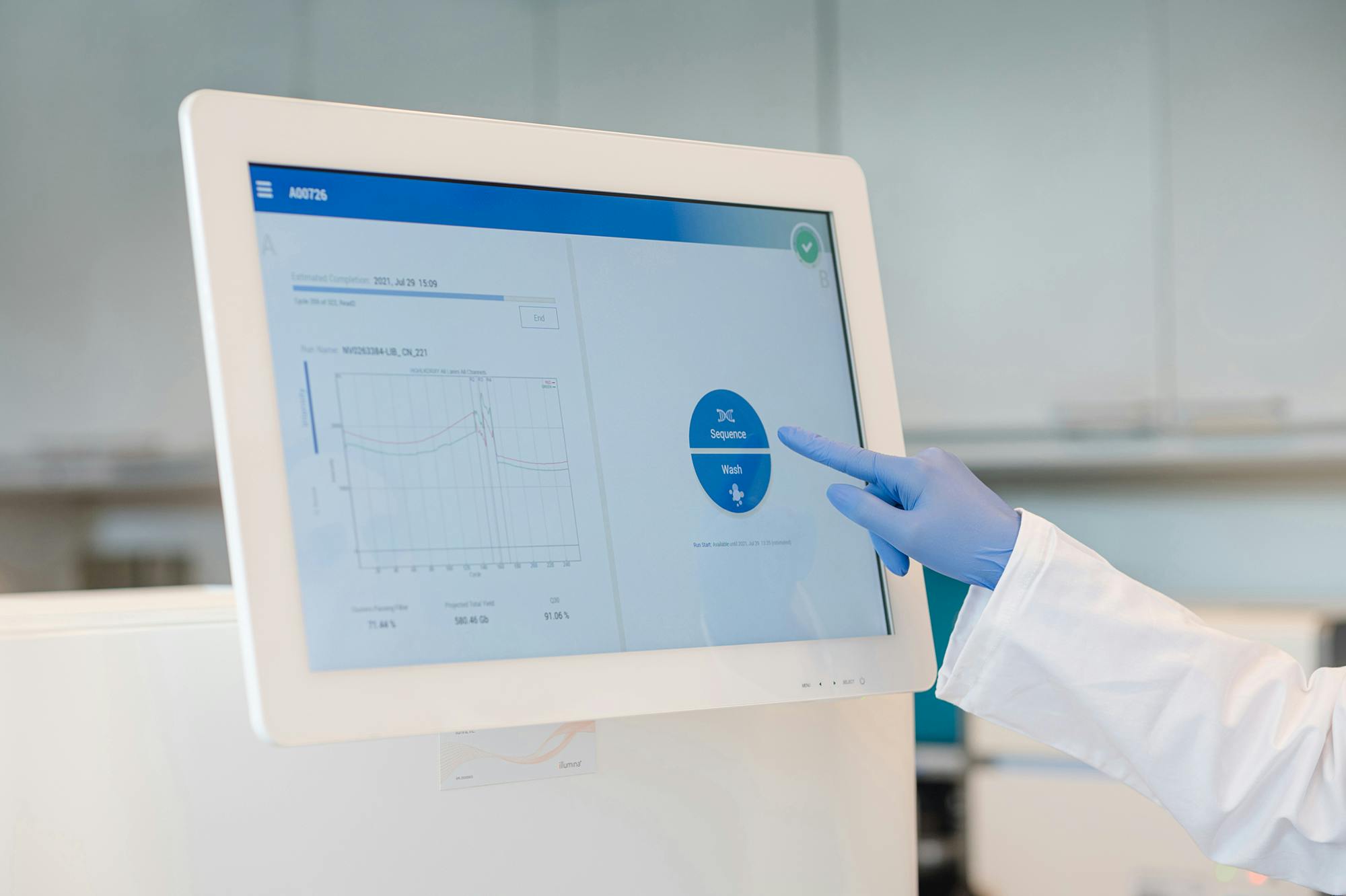
Medicover Genetics bietet ein umfangreiches Portfolio an genetischen Tests an, darunter zytogenetische Analysen, molekularpathologische Lösungen, die neueste Next Generation Sequencing (NGS)-Technologie sowie die Sequenzierung des Mikrobioms, um medizinischen Fachkräften zu helfen, die am besten geeigneten genetischen Tests für ihre Patient:innen zu finden.
In unseren genetischen Beratungsstellen bieten wir umfassende genetische Beratungsdienste an, um Patient:innen und Familien während des Testprozesses zu begleiten. Unser Team zertifizierter medizinisch-genetischer Berater:innen bietet einfühlsame Unterstützung und hilft Patient:innen, auf der Grundlage ihrer genetischen Daten, fundierte Entscheidungen über ihre Gesundheit und ihr Wohlbefinden zu treffen.
Als Ihr Laborpartner bieten wir in Fachbereichen wie der Endokrinologie, Gynäkologie & Reproduktionsmedizin, Inneren Medizin, Kardiologie und Transfusionsmedizin eine fachübergreifende Diagnostik, die genetische Diagnostik und klassische Labormedizin kombiniert. Diese interdisziplinäre Herangehensweise ermöglicht es uns, komplexe diagnostische Fragestellungen präzise und fundiert zu beantworten und Ihnen somit einen entscheidenden Mehrwert in der Patientenversorgung zu bieten. Unsere Mission: Sie in Ihrer medizinischen Expertise optimal zu unterstützen – präzise, fundiert und interdisziplinär.

Die Whole Exome Sequenzierung (WES) ist ein umfassender genetischer Test, der es ermöglicht, die proteinkodierenden Bereiche (Exons) aller menschlichen Gene (etwa 20.000 Gene) zu analysieren. Die Gesamtheit aller Exons wird als Exom bezeichnet. Das menschliche Exom enthält etwa 85% der bekannten, krankheitsverursachenden Varianten. Durch den Einsatz von WES können in einem Ansatz alle Gene auf das Vorliegen von Varianten untersucht werden, die im Zusammenhang mit der Symptomatik des Patienten stehen könnten.

Seit 1995 setzen wir Maßstäbe in der Gesundheitsversorgung und Diagnostik. Werden Sie Teil unserer Mission, exzellente Medizin in 14 Ländern rund um den Globus anzubieten. Bei Medicover steht der Mensch im Mittelpunkt. Wir bieten nicht nur unseren Patient:innen die beste Betreuung, sondern auch unseren Mitarbeiter:innen. Entfalten Sie Ihr volles Potential in einem Umfeld, das Ihre Entwicklung und Weiterbildung aktiv fördert. Besuchen Sie unsere Karriereseite und finden Sie heraus, wie Sie Teil unserer leidenschaftlichen Medicover-Familie werden können.

