Gastrointestinale Stromatumoren (GIST)
Gastrointestinale Stromatumoren (GIST) sind die häufigsten mesenchymalen Neoplasien des Magen-Darm-Traktes, die morphologisch in spindelzellige, epitheloide und gemischte Subtypen unterteilt werden. Immunhistochemisch zeigen etwa 95% der GIST eine Positivität für CD117 (KIT-Rezeptor) oder DOG1. Die meisten GIST weisen genetische Varianten in den Genen KIT oder PDGFRA auf, die für Rezeptor-Tyrosinkinasen kodieren und bei Mutationen zu einer konstitutiven Aktivierung von Zellproliferation führenden Signalwegen führen. Die Behandlung umfasst in der Regel den Einsatz von Tyrosinkinaseinhibitoren wie Imatinib, wobei die genetische Analyse entscheidend für die Prognose und das Ansprechen auf die Therapie ist.
Wissenschaftlicher Hintergrund
GIST sind insgesamt seltene Tumoren, jedoch die häufigsten mesenchymalen Neoplasien des gastrointestinalen Traktes. Die häufigsten Primärlokalisationen liegen im Bereich des Magens (50-60%) und des Dünndarms (20-30%), seltener im Kolorektum (5-10%) und Ösophagus (<1%). GIST repräsentieren ein morphologisches und biologisches Kontinuum von zufällig entdeckten, <10 mm benignen Mikro-GIST bis zu großen Sarkomen. Die GIST werden in 3 morphologische Untergruppen eingeteilt: ca. 70% sind vom spindelzelligen Subtyp, ca. 10 % sind vom epithelioiden Subtyp und weitere 20% sind vom gemischt spindelzellig-epithelioiden Subtyp. Zur histologischen Diagnosesicherung eines GIST ist die Immunhistochemie unerlässlich. In etwa 95% aller GIST ist der Antikörper gegen CD117 (KIT-Rezeptor) oder DOG1 nachweisbar. Etwa 70-80% der Fälle exprimieren zudem das stammzellnahe Antigen CD34.
Trotz der klinikopathologischen Unterschiede teilen die meisten GIST das gleiche genetische Profil. Dieses beinhaltet KIT- und PDGRFA-Varianten, welche sich gegenseitig ausschließen. Beide Gene codieren für Rezeptor-Tyrosinkinasen, die nach Bindung eines Liganden Signalwege aktivieren und somit wichtige Zellfunktionen wie Zellproliferation oder Apoptose steuern. Durch Varianten in KIT und PDGRFA kommt es zu einer konstitutiven Aktivierung der Tyosinkinasen. Diese wird bereits in kleinen Tumoren mit einem Durchmesser <1 cm nachgewiesen, so dass es sich um ein frühes pathogenetisches Ereignis handelt. Da die genetische Analyse sowohl für die prognostische Bedeutung als auch die Prädiktion des Therapieansprechens relevant ist, sollte sie bereits zum Zeitpunkt der Erstdiagnose angestrebt werden.
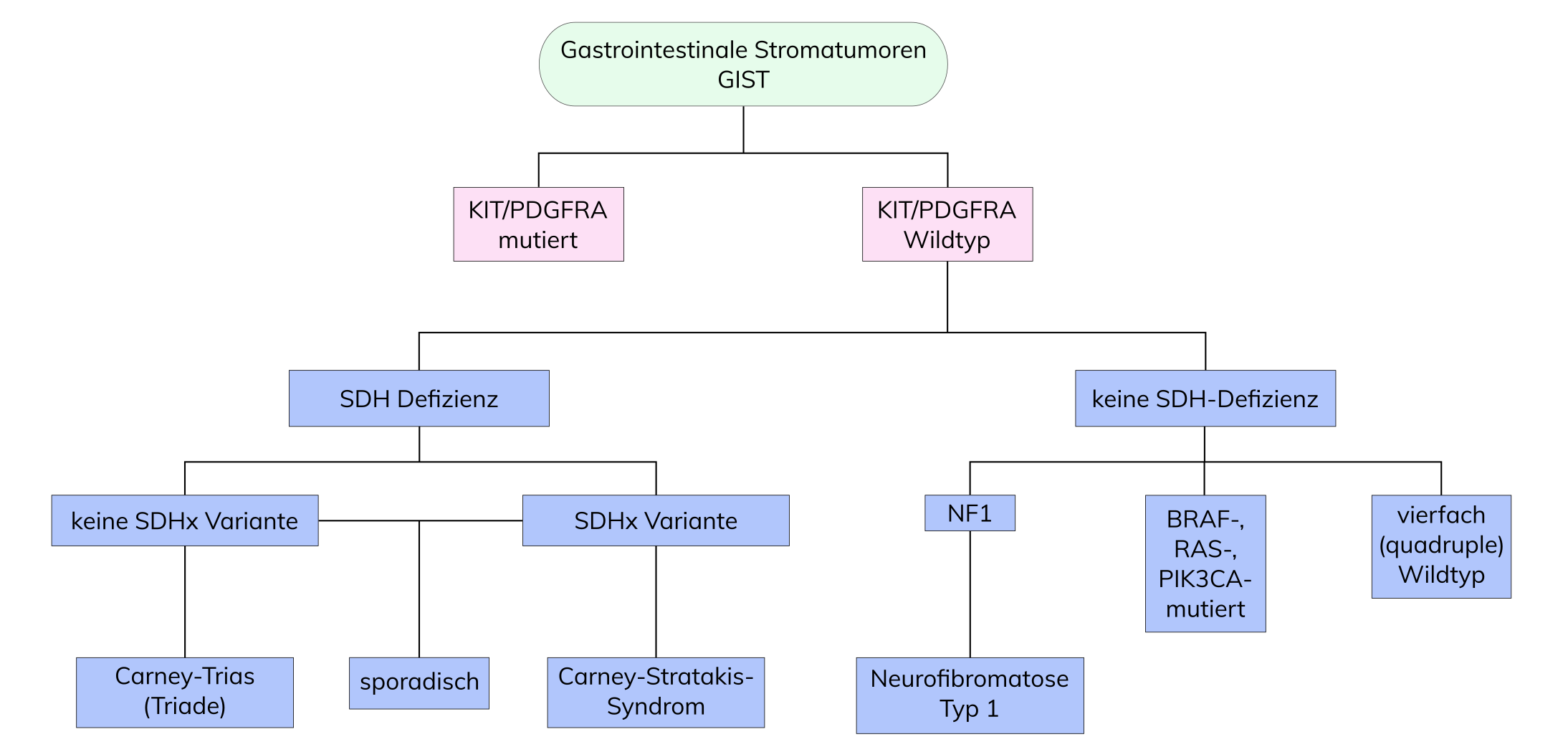
Eine hereditäre Prädisposition wird nur sehr selten beobachtet. Dabei handelt es sich entweder um familiäre GIST mit entsprechender Keimbahnvariante in KIT, um GIST im Kontext des Carney-Strakatis-Syndroms oder um GIST im Zusammenhang mit der Neurofibromatose-1 (Morbus von Recklinghausen).
Die Häufigkeit von KIT-Varianten liegt bei etwa 80-90%. Patienten mit Varianten im unbehandelten Primärtumor sprechen in der Regel auf eine Therapie mit dem Tyrosinkinaseinhibitor Imatinib an. Die häufigsten Varianten in KIT finden sich in Exon 11 (60%) und sind hinsichtlich Länge und Typ (Deletionen, Insertionen, Punktmutationen und Kombinationen) sehr heterogen. Patienten mit einer KIT-Exon 11-Deletion weisen ein höheres Rezidivrisiko auf als solche mit Exon 11-Insertion oder -Punktmutation, PDGFRA-Variante oder Wildtyp. In 10-15% der Fälle finden sich KIT Varianten in Exon 9. Hier tritt fast immer die gleiche 6-Basenpaar-Insertion auf, die zu einer Duplikation der Aminosäuren Alanin 502 und Tyrosin 503 führt. Exon 9-Varianten kommen prädominant in GIST mit Lokalisation im Dünndarm vor. Sie zeigen nur in etwa der Hälfte der Fälle ein Ansprechen auf Imatinib. Es konnte jedoch gezeigt werden, dass diese Patienten davon profitieren, wenn sie von Beginn an mit 800 mg Glivec/Tag statt 400 mg Glivec/Tag oder mit einem anderen TKI behandelt werden. Zusätzlich sind Varianten beschrieben, die während einer Behandlung mit einem TKI detektiert werden und zu einer Resistenz führen. Weitere KIT-Varianten finden sich selten u.a. in Exon 13 (Lys642Glu) und Exon 17 (Asn822Lys). Bei KIT-Exon 13- und 14-Varianten ist eine Therapie mit dem Zweitlinien-TKI Sunitinib möglich. Bei der Exon 17-Variante (Asn822Lys) ist eine primäre Resistenz zu erwarten. Bei sekundären KIT-Exon 17- und 18-Varianten ist auch Sunitinib in der Regel unwirksam. Es konnte ein Ansprechen auf Sorafenib und Regorafenib gezeigt werden.
Die Häufigkeit von PDGFRA-Varianten liegt bei etwa 10-15%. Auch hier sprechen Patienten mit Varianten im unbehandelten Primärtumor in der Regel auf eine Therapie mit dem Tyrosinkinaseinhibitor Imatinib an. Findet sich jedoch die Variante Asp842Val, so ist diese mit einer Resistenz gegenüber Imatinib assoziiert. Bei diesen Tumoren ist ebenso eine Therapie mit einem Zweitlinien-TKI Sunitinib oder Drittlinien-TKI Regorafenib indiziert.
GIST, die keine Varianten in KIT oder PDGFRA aufweisen, werden in Succinatdehydrogenase (SDH)-defiziente und nicht-SDH-defiziente GIST klassifiziert. Die SDH-defiziente Gruppe macht 20-40-% der KIT/PDGFRA-Wildtyp GIST aus und ist gekennzeichnet durch einen Verlust der Expression der SDH-Untereinheit B (SDHB), meist durch sporadische und/oder Keimbahnvarianten in SDHA, SDHB, SDHC und SDHD (SDHx). Zur SDH-defizienten Gruppe zählen neben sporadischen Fällen die Carney Trias und das Carney-Strakatis-Syndrom. Die nicht-SDH-defiziente Gruppe beinhaltet Neurofibromatose Typ 1 mit Varianten in NF1 sowie GIST mit Varianten BRAF, KRAS und PIK3CA.
Li et al. 2019, Onco Targets Ther 12:5123 / Wada et al. 2016, Pathol Int 66:431 / Nannini et al. 2017, J Transl Med 15:113 / Cioffi et al. 2015, J Clin Oncol 33:1849 / Gheorghe et al. 2014, J Med Life 7:139 / Reichardt et al. 2022, Gastrointestinale Stromatumoren (GIST). Onkopedia
letzte Aktualisierung: 3.11.2023