Mastozytose
Mastozytose ist eine klonale Mastzellerkrankung mit einer variablen klinischen Präsentation, die von kutanen Formen bis hin zu aggressiven systemischen Varianten mit Multiorganbeteiligung reicht. Die Diagnose erfordert den Nachweis von Mastzellinfiltraten sowie weitere molekulare Kriterien, einschließlich aktivierender Varianten im KIT-Gen. Die WHO klassifiziert Mastozytose in verschiedene Subtypen, wobei die systemische Mastozytose (SM) die häufigste Form bei Erwachsenen ist und in weitere Untergruppen unterteilt wird. Die Behandlungsoptionen umfassen den Einsatz von Tyrosinkinaseinhibitoren, wobei Resistenzmechanismen und die Entwicklung selektiverer Inhibitoren Herausforderungen darstellen.
Wissenschaftlicher Hintergrund
Die Mastozytose ensteht durch eine klonale, neoplastische Proliferation von Mastzellen mit Infiltration eines oder mehrerer Organsysteme. Die klinische Präsentation ist heterogen und reicht von rein kutaner Manifestation und Spontanremissionen bis hin zu aggressiven Verläufen mit Multiorganversagen und stark eingeschränkter Lebenserwartung. Krankheitssymptome resultieren aus einer Infiltration der Organe durch die klonalen Mastzellen und der damit verbundenen Freisetzung von Mastzellmediatoren (v.a. Histamin, Prostaglandine, Leukotriene).
Prinzipiell handelt es sich um eine erworbene klonale Erkrankung, familiäre Fälle sind aber beschrieben. Mastozytosen können vom Säugling bis zum Erwachsenen alle Altersstufen betreffen. Bei den meisten pädiatrischen Patienten ist die Erkrankung auf die Haut mit unterschiedlichen Läsionen begrenzt und wird als kutane Mastozytose (CM) bezeichnet. Sie hat eine Tendenz zur Spontanremission. Bei den meisten erwachsenen Patienten hingegen handelt es sich um eine systemische Mastozytose (SM) mit Beteiligung zumindest eines extrakutanen Organs – wobei prinzipiell jedes Organ einbezogen sein kann – häufig mit einer Infiltration des Knochenmarks. Daher ist eine Knochenmarkuntersuchung bei Verdacht auf systemische Mastozytose bei Erwachsenen nachdrücklich zu empfehlen.
Differentialdiagnostisch entscheidend ist die Abgrenzung gegenüber einer Mastzellenhyperplasie und Mastzellaktivierung anhand charakteristischer morphologischer und molekularer Kennzeichen.
Die aktuelle WHO-Klassifikation unterscheidet drei klinische Formen, wobei die systemische Mastozytose noch in 5 Untergruppen unterteilt wird:
1. Kutane Mastozytose (CM)
2. Systemische Mastozytose
- Indolente systemische Mastozytose (ISM)
- “Smouldering” systemische Mastozytose (SSM)
- Systemische Mastozytose, assoziiert mit hämatologischer Neoplasie (SM-AHN)
- Aggressive systemische Mastozytose (ASM)
- Mastzellleukämie (MCL)
3. Mastzellsarkom
Bei den meisten Patienten wird eine indolente systemische Mastozytose (ISM) diagnostiziert. Aggressivere Untergruppen wie aggressive SM (ASM) und Mastzellleukämie (MCL) sind selten. Etwa 5-20% der Patienten mit SM zeigen oder entwickeln eine systemische Mastozytose assoziiert mit hämatologischer Neoplasie (SM-AHN), welche die zweithäufigste Form einer SM darstellt.
Die klinische Diagnose SM erfordert entweder ein Hauptkriterium und ein Nebenkriterium oder ≥ 3 Nebenkriterien:
Hauptkriterium:
Mastzellinfiltrate (> 15 Mastzellen) im Knochenmark und/oder anderen extrakutanen Organen
Nebenkriterien
- Nachweis atypischer Mastzellen im befallenen Gewebe
- Nachweis einer aktivierenden Variante im KIT-Gen im Knochenmark, Blut oder einem anderen extrakutanen Organ
- Nachweis einer CD25-Expression mit oder ohne zusätzlicher CD2-Expression auf den neoplastischen Mastzellen
- persistierende Serum-Tryptase > 20ng/ml (wenn keine zusätzliche myeloide Neoplasie vorliegt bzw. Anpassung bei Vorliegen einer hereditären Alpha-Tryptasämie)
Diagnostik
Zu den molekulargenetisch-diagnostischen Kriterien der SM gehört der Nachweis einer Variante im KIT-Gen. Die KIT Asp816Val-Variante findet sich in >90% der Patienten mit SM, während andere aktivierende Varianten in der extrazellulären (Deletion von Codon 419 in Exon 8, Ala502_Tyr503dup in Exon 9), Transmembran- (Phe522Cys) oder Juxtamembran-Domäne (Val560Gly) in etwa <1% der fortgeschrittenen SM Fälle und häufiger bei indolenter SM nachgewiesen werden. Sie führen zu einer Mastzellaktivierung und -proliferation durch die Bindung des Stammzellfaktors SCF an den KIT-Rezeptor und somit zu einer Liganden-unabhängigen Aktivierung dieses Signalwegs. KIT-Varianten alleine können allerdings nicht die verschiedenen klinischen Manifestationen der Erkrankung erklären. Gerade bei der ISM ist der Anteil der neoplastischen Mastzellen im Knochenmark sehr gering, weshalb sehr sensitive Techniken wie Droplet Digital PCR (ddPCR) zum Nachweis der KIT Asp816Val-Variante zum Einsatz kommen sollten.
Eine Knochenmark-Mastozytose ist ein separater Subtyp der SM und charakterisiert durch die Abwesenheit von Hautläsionen, B-Findings (Nachweis einer KIT Asp816Val-Variante mit VAF >=10% im Knochenmark oder peripheren Blutleukozyten qualifiziert als B-Finding) und basale Serumtryptase <125 ng/ml.
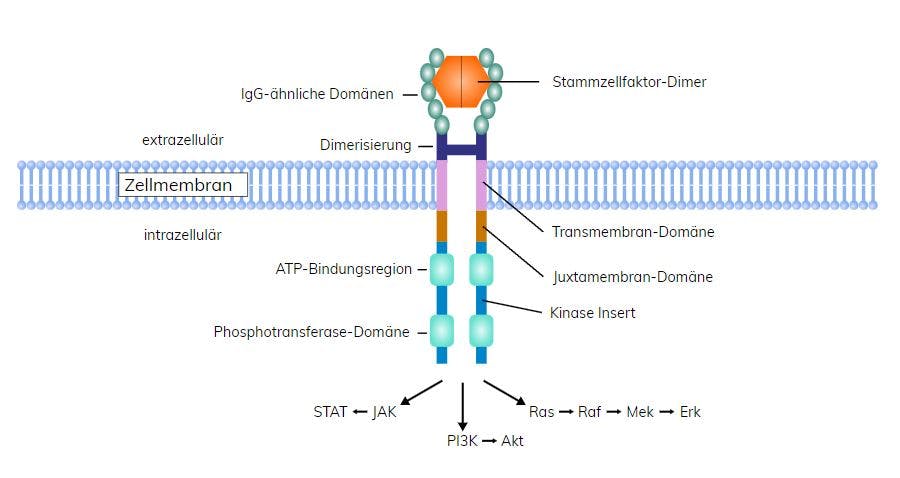
Die konstitutive Aktivierung von KIT bietet prinzipiell die Möglichkeit einer modernen zielgerichteten Therapie mit Tyrosinkinaseinhibitoren. Allerdings induzieren die häufigen KIT Asp816Val/His/Tyr/Asn-Varianten eine Resistenz gegenüber den bei anderen myeloischen und soliden Neoplasien etablierten Tyrosinkinaseinhibitoren (z.B. Imatinib, Nilotinib und Masitinib). Lediglich der Breitspektrum- bzw. Multikinaseinhibitor Midostaurin zeigt zufriedenstellende Aktivität gegenüber KIT Asp816 und führt zu Ansprechraten von bis zu 60%. Er ist die derzeit einzige zugelassene zielgerichtete Therapie bei fortgeschrittener SM. Weitere, selektivere TKI sind in klinischer Erprobung, so z.B. Avapritinib, das sowohl an KIT Asp816 als auch an PDGFRα bindet. Dieses weist vorläufigen klinischen Ergebnissen zufolge eine stärkere Ansprechrate auf, womöglich aber auch gravierendere Nebenwirkungen.
In den letzten Jahren konnten zudem eine Reihe von Varianten in anderen Genen nachgewiesen werden, die den SM-Phänotyp modifizieren und/oder zur Krankheitsprogression beitragen können. Varianten in TET2 finden sich in etwa 29% der SM-Fälle (15% ISM, 40% ASM, 35% SM-AHN), wobei in 50% dieser Patienten die TET2-Variante mit einer KIT Asp816Val-Variante kosegregiert und dessen onkogenes Potential erhöhen kann. Varianten in TET2 sind mit einem schlechteren Gesamtüberleben assoziiert. In 17% der SM-Patienten finden sich Varianten in ASXL1 sowie in 11% der SM-Patienten in CBL. Dabei ist die Varianten-Allel-Frequenz (VAF) von SM-AHN über ASM zu ISM abnehmend. Bei einer fortgeschrittenen SM sind Varianten in ASXL1, CBL und RUNX1 mit einem schlechteren Gesamtüberleben assoziiert, unabhängig vom Alter und WHO-Subtyp.
Khoury et al. 2022, Leukemia, 36:1703 / Gillreath et al. 2019, Clin Pharm 11:77 / Swerdlow, Campo, Harris, Jaffe, Pileri, Stein, Thiele (Eds), 2017, WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition), Chapter 3:62 / Arber et al. 2016, Blood 127:2391 / Pardanani et al. 2016, Am J Hematol 91:888 / Arock et al. 2015, Leukemia 29:1223 / Pardanani et al. 2015, Br J Haematol 175:534 / Chatterjee et al. 2015, Oncotarget 6:18250
letzte Aktualisierung: 3.11.2023