Hereditäre Alpha-Tryptasämie (HaT)
Die Hereditäre Alpha-Tryptasämie (HaT) ist eine autosomal-dominante Erkrankung mit einer Prävalenz von 4-6% in der westlichen Bevölkerung, die durch Duplikationen oder Triplikationen des TPSAB1-Gens verursacht wird und zu erhöhten basalen Serumtryptase (BST)-Werten führt. Trotz vollständiger Penetranz variiert die Expressivität, sodass Symptome und deren Schweregrad individuell unterschiedlich ausfallen können. HaT zeigt Überschneidungen mit Mastzellerkrankungen wie systemischer Mastozytose (SM). Die Diagnose von HaT kann invasive Untersuchungen reduzieren und ist entscheidend für die Bewertung und Behandlung von Mastzellerkrankungen.
Wissenschaftlicher Hintergrund
Die Hereditäre Alpha-Tryptasämie (HaT) findet sich mit einer Prävalenz von 4-6% in der westlichen Bevölkerung und ist ein autosomal-dominant vererbtes genetisches Merkmal, das durch erhöhte Keimbahnkopien vom Gen TPSAB1, welches für Alpha-Tryptase codiert, verursacht wird. Dies führt zu einer erhöhten basalen Serumtryptase (BST). Die Erkrankung ist nahezu vollständig penetrant, jedoch mit unterschiedlicher Expressivität, so dass Personen mit HaT einheitlich erhöhte BST-Werte (fast überall >8ng/mL), aber unterschiedliche damit assoziierte Phänotypen aufweisen. Die Symptome können sich in jedem Alter manifestieren, wobei etwa ein Drittel der ansonsten gesunden Erwachsenen mit HaT signifikante Symptome, ein Drittel leichte bis mittelschwere Symptome und ein Drittel Symptome mit der gleichen Häufigkeit wie Personen ohne HaT aufweisen. Es ist eine Vielzahl von Symptomen beschrieben, jedoch sollte eine TPSAB1-Genotypisierung bei Patienten mit folgenden Symptomen erfolgen: einer Mastzellaktivierung, Anaphylaxie in der Vorgeschichte, gastrointestinalen Symptomen, die mit nächtlichem Erwachen verbunden sind, oder Darmentzündungen, die nicht auf die Therapie ansprechen oder atypisch sind, sowie BST-Werten >8ng/mL.
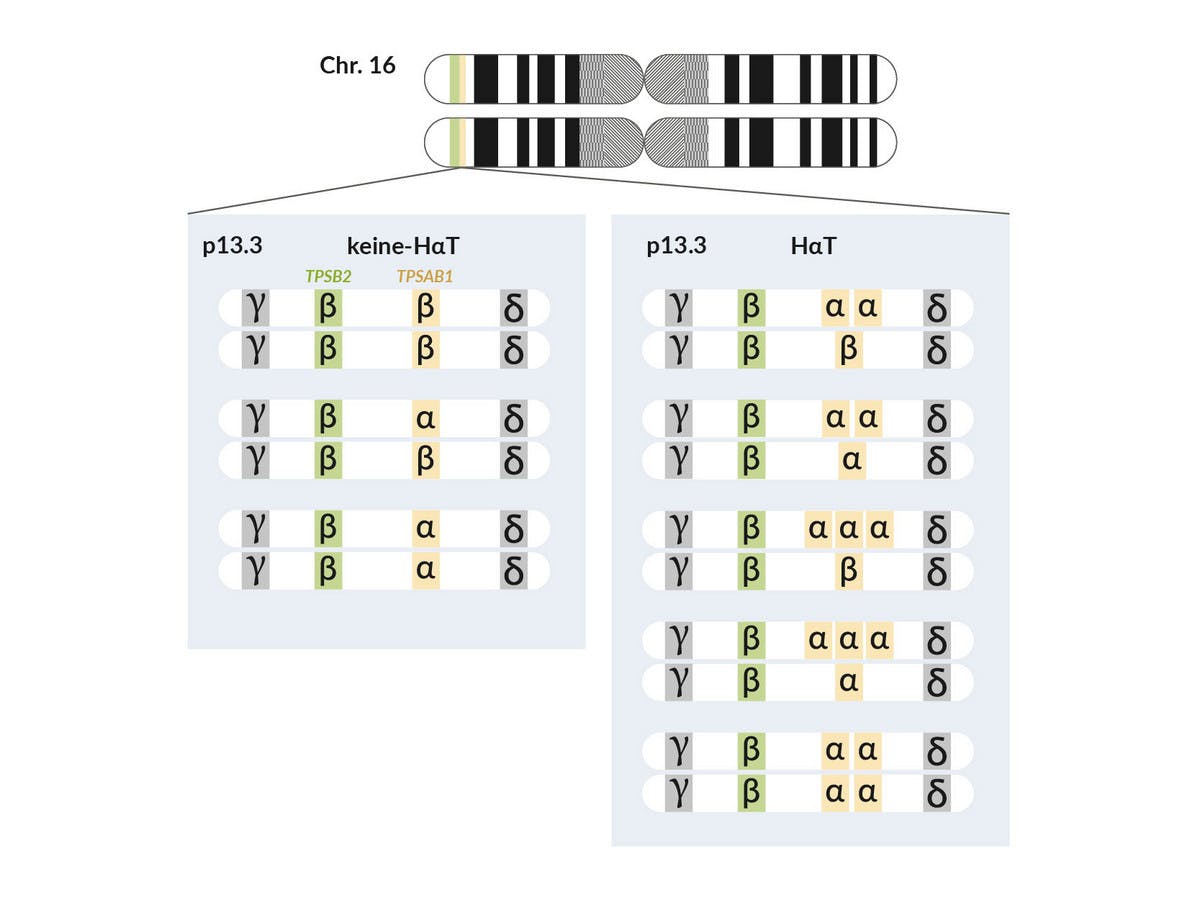
Der Tryptase-Locus enthält vier für Tryptase codierende Gene (TPSG1, TPSB2, TPSAB1 und TPSD1) und liegt in der Chromosomenregion 16p13.3. Die sekretierten Formen der Tryptase, die von klinischen Laboren gemessen und als BST-Wert angegeben werden, codieren jedoch nur TPSB2 und TPSAB1. Während angenommen wird, dass TPSB2 nur Beta-Tryptase-Isoformen codiert, codiert der TPSAB1-Locus entweder Alpha- oder Beta-Isoformen. Lange wurde angenommen, dass Individuen jeweils eine Kopie von TPSB2 und TPSAB1 haben. Basierend auf der Expression der Tryptase-Isoform an diesen zwei genetischen Loci wurden in der Literatur drei kanonische Genotypen beschrieben, die nicht mit einer HaT einhergehen: ββ:ββ , βα:ββ oder βα:βα (beide Alpha-Tryptase-Kopien auf unterschiedlichen Allelen). Bei erhöhtem BST-Wert kann eine erhöhte TPSAB1-Kopienzahl in Form von Duplikationen oder Triplikationen auf einem Allel vorliegen; dies wird dann als HaT klassifiziert. Dabei besteht ein Gendosiseffekt zwischen der Anzahl zusätzlicher TPSAB1-Kopien, dem BST-Wert und dem Schweregrad der klinischen Symptome.
Tryptasen werden fast ausschließlich von Mastzellen (MC) freigesetzt. Basophile, der einzige andere Zelltyp, von dem bekannt ist, dass er bei gesunden Personen α- und β-Tryptase absondert, produzieren allerdings nur <1% der Tryptase, die MCs produzieren.
Beobachtungen zeigen, dass zahlreiche klinische Ähnlichkeiten zwischen Personen mit HaT und Personen mit klonalen und nicht-klonalen Mastzell(MC)-Erkrankungen, einschließlich MC-Aktivierungssyndrom (MCAS) und systemischer Mastozytose (SM) bestehen. So zeigt sich eine erhöhte Prävalenz von HaT bei diesen Patienten, was mit einer häufigeren und/oder schwereren Anaphylaxie sowie vermehrten Mastzellmediator-assoziierten Symptomen bei SM verbunden ist. Etwa 12-17% der Patienten mit SM haben ebenfalls eine HaT. Daher findet sich in einem Vorschlag zur Anpassung der WHO-Kriterien zur Diagnose SM u.a. eine Einschränkung zum BST-Wert. Der BST-Wert im Serum ist bei den meisten SM-Patienten erhöht. Daher ist ein deutlich erhöhter BST-Wert im Serum ein untergeordnetes SM-Kriterium. Der Konsensschwellenwert liegt bei 20 ng/mL. Bei Patienten mit bekannter HaT sollte der BST-Wert jedoch korrigiert werden. Ein vorgeschlagener Ansatz besteht darin, den BST durch eins plus die Anzahl zusätzlicher Alpha-Tryptase-Genkopien zu dividieren, so dass dieser als SM-Kriterium ggf. wegfällt. Hier fehlen aber noch ausreichende Validierungsstudien.
Tabelle: Tryptase-Genotpyen und assoziierte BST-Werte (modifiziert nach Glover et al. 2021, Ann Allergy Asthma Immunol, 127:638)
| Tryptase-Genotpyen und assoziierte BST-Werte, beeinflußt durch zusätzliche TPSAB1-Kopien (codiert für Alpha-Tryptase) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Zusätzliche TPSAB1-Kopien | 0 | 1 | 2 | 3 | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tryptase-Genotypena (TPSAB1, TPSB2) | β,β/β,β α,β/β,β α,β/αβ α,β/β,β,β β,β/β,β,β β/α,β β/β,β | αα,β/β,β αα,β/α,β αα,ββ/ββ αα,ββ/αβ | αα,β/αα,β ααα,β/β,β ααα,β/α,β | ααα,β/αα,β | αααα,β/β,β αααα,β/α,β | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die Feststellung einer HaT-Diagnose kann den Bedarf an weiterer invasiver und umständlicher Aufarbeitung, einschließlich Gewebe- und/oder Knochenmarkbiopsien, bei Personen reduzieren, die ansonsten keine Anzeichen und Symptome aufweisen, die auf klonale Mastzellen oder andere myeloische Dyskrasien hindeuten. Darüber hinaus sollte der Nachweis einer erhöhten BST ohne HaT oder eine fortgeschrittene Nierenerkrankung eine klinische Evaluation für eine klonale Erkrankung innerhalb des myeloischen Kompartiments nach sich ziehen.
Luskin et al. 2021, J Allergy Clin Immunol Pract 9:2235 / Glover et al. 2021, Ann Allergy Asthma Immunol 127:638 / Valent et al. 2021, Hemasphere 5:e646 / Lyons 2018, Immunol Allergy Clin North Am 38:483
letzte Aktualisierung: 3.11.2023