X-Inaktivierung
Die X-Inaktivierung ist ein entscheidender Mechanismus zur Dosiskompensation von X-gebundenen Genen zwischen den Geschlechtern, wobei in weiblichen Zellen eines der beiden X-Chromosomen zufällig inaktiviert wird. Dieser Prozess wird durch das X inactivation center (Xic) und die Expression der nicht codierenden Xist-RNA initiiert. In der Humangenetik beeinflusst die X-Inaktivierung die Ausprägung von X-gebundenen Erkrankungen, wobei ungleiche oder verschobene Inaktivierungsmuster den Schweregrad der Symptome beeinflussen können. Metabolische Kooperation zwischen Zellen kann Defekte bei Trägerinnen von X-gebundenen Erkrankungen kompensieren, während zelluläre Interferenz in bestimmten Fällen zu schwereren Phänotypen bei Frauen führen kann.
Wissenschaftlicher Hintergrund
Bereits 1949 wurde eine dichte Struktur im Zellkern von weiblichen somatischen Zellen entdeckt (Barr Körperchen), die 1960 als inaktiviertes X-Chromosom identifiziert wurde. Die Genetikerin Mary Frances Lyon stellte im darauffolgenden Jahr die Hypothese auf, dass in weiblichen Zellen eines der beiden X-Chromosomen dauerhaft inaktiviert wird, um zu verhindern, dass weibliche Zellen doppelt soviele X-gebundene Gene exprimieren wie männliche Zellen (Lyon Hypothese). Das X-Chromosom trägt über 1000 Gene und liegt bei Männern in nur einfacher Ausführung vor. Frauen tragen in jeder Zelle zwei X-Chromosomen und würden im Vergleich zu Männern somit die doppelte Gendosis aufweisen. Die zufällige Inaktivierung des entweder paternalen (väterlichen) X-Chromosoms (Xp) oder maternalen (mütterlichen) X-Chromosoms (Xm) bei Frauen dient der Dosiskompensation von X-gebundene Genen zwischen beiden Geschlechtern, so dass weibliche Zellen die gleiche Gendosis aufweisen wie männliche Zellen mit nur einem X-Chromosom. Durch die zufällige Inaktivierung eines der beiden X-Chromosomen stellen Frauen ein Mosaik aus zwei Zellpopulationen dar, Zellen mit aktivem Xm oder aktivem Xp.
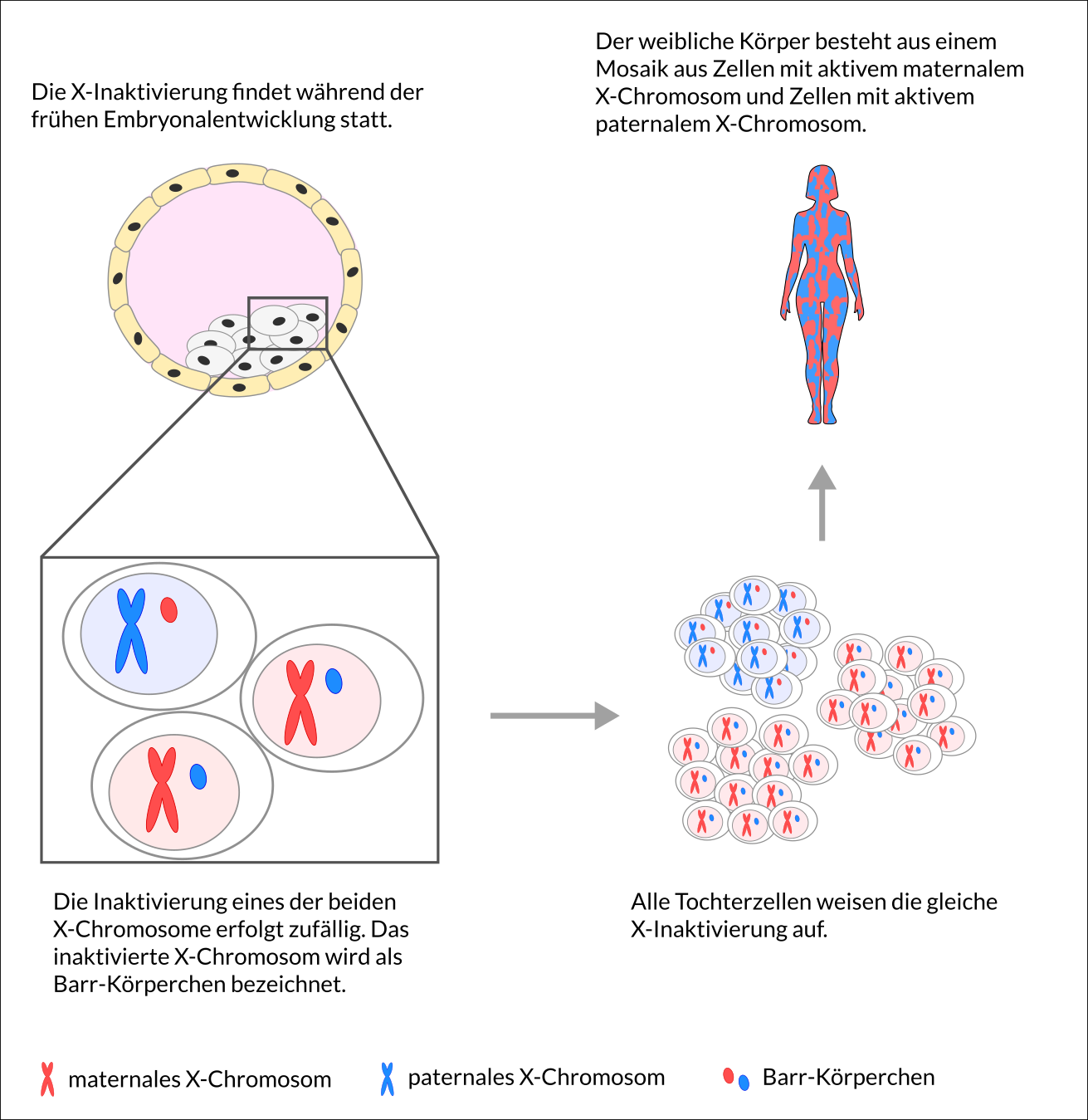
Die X-Inaktivierung erfolgt in der frühen weiblichen Embryonalentwicklung und unterliegt einem sehr komplexen molekularen Mechanismus, der bis heute nicht vollständig aufgeklärt ist. Für die X-Inaktivierung muss eine Zelle verschiedene Aufgaben erfüllen können:
- Bestimmung der Anzahl der X-Chromosomen
- Unterscheidung der X-Chromosomen
- Auswahl des zu inaktivierenden X-Chromosoms
- Prozess der Inaktivierung
Eine essentielle Komponente bei der X-Inaktivierung ist eine Region auf dem X-Chromosom, die als X inactivation center (Xic) bezeichnet wird. Durch Experimente hat man herausgefunden, dass die X-Inaktivierung nur erfolgt, wenn in der Zelle zwei (oder mehr) Xic vorliegen. Im Xic liegt das Xist (X inactivation specific transcript)-Gen, das für eine nicht codierende RNA codiert (Xist-RNA). In der frühen weiblichen Embryonalentwicklung wird die Xist-RNA in den Zellen nur von einem der beiden X-Chromosomen exprimiert. Die Xist-RNA verbleibt im Zellkern und überzieht dort das Chromosom, von dem sie exprimiert wird, wodurch die Inaktivierung des gesamten Chromosoms induziert wird.
Die X-Inaktivierung ist ein Prozess, der in der Regel immer initiert wird, sobald zwei oder mehr X-Chromosomen in der Zelle vorhanden sind, mit dem Ziel, dass nur ein aktives X-Chromosom in der Zelle verbleibt. Daher zeigt sich auch bei Männern mit dem Genotyp XXY (Klinefelter Syndrom) die Inaktivierung eines der beiden X-Chromosomen, und bei Frauen mit dem Genotyp XXX (Triplo-X-Syndrom) kann die Inaktivierung von zwei der insgesamt drei X-Chromosomen beobachtet werden. Es gibt einige Gene auf dem inaktivierten X-Chromosom, die der allgemeinen Inaktivierung entkommen (ca. 15%). Daher zeigen sich bei den oben genannten Syndromen trotz der X-Inaktivierung der überzähligen X-Chromosomen verschiedene Symptome, die auf die abweichende Dosis dieser Gene zurückgeführt werden können.
In der Humangenetik spielt die X-Inaktivierung eine besondere Rolle bei der Ausprägung von X-chromosomal vererbten Erkrankungen. Bei X-gebunden rezessiven Erkrankungen kann folgendes beobachtet werden:
- In der Regel sind nur Männer betroffen, da diese nur ein X-Chromosom tragen.
- Bei einer Frau kann in sehr seltenen Fällen eine X-rezessive Erkrankung auftreten, wenn sie auf beiden X-Chromosomen ein mutiertes Allel geerbt hat oder wenn die X-Inaktivierung zugunsten des mutationstragenden X-Chromosoms verschoben ist.
- Väter können die Erkrankung nicht an Söhne vererben. Alle Töchter sind heterozygote Trägerinnen (Konduktorinnen).
- Nachkommen einer Konduktorin erben die Mutation mit einer Wahrscheinlichkeit von 50%, Mädchen mit der Mutation sind damit wiederum Konduktorinnen, Jungen mit der Mutation erkranken;
Bei Trägerinnen einer X-gebunden rezessiven Erkrankung wird aufgrund der zufälligen X-Inaktivierung nur in ca. 50% der Zellen das mutierte rezessive Allel exprimiert. Die andere Hälfte der Zellen exprimiert das funktionelle Allel, wodurch häufig eine Kompensation des ererbten Defekts möglich ist und sich somit bei Frauen in der Regel phänotypisch keine oder eine mildere Symptomatik zeigt. Durch Zell-zu-Zell-Verbindungen oder über Endozytose werden Metabolite bzw. Enzyme zwischen Zellen ausgetauscht, wodurch der Defekt in den mutierten Zellen kompensiert wird. Diese Kompensation wird auch als metabolische Kooperation (metabolic cooperation) bezeichnet und kann bei verschiedenen Erkrankungen beobachtet werden:
Morbus Fabry
Morbus Fabry ist eine lysosomale Speicherkrankheit, bei der das Enzym alpha-Galactosidase A einen Defekt aufweist und es dadurch zu einer Akkumulation von Glycosphingolipiden in verschiedenen Geweben und Organen kommt. Da die alpha-Galactosidase A von nicht betroffenen Zellen zu mutierten Zellen transferiert werden kann, zeigen heterozygote Trägerinnen in der Regel nur eine milde Symptomatik.
Morbus Hunter
Beim Morbus Hunter kommt es aufgrund eines Defekts der Iduronat-Sulfatase im Körper zu einer Akkumulation von Mucopolysacchariden. Bei heterozygoten Trägerinnen können auch hier die Zellen, die das funktionelle Allel exprimieren, den Defekt durch den Transfer des Enzyms zu mutierten Zellen kompensieren, so dass Frauen entweder nur sehr geringe oder gar keine Symptome zeigen.
Lesch-Nyhan Syndrom
Dem Lesch-Nyhan Syndrom liegt eine Hypoxanthin-Phosphoribosyltransferase (HPRT) Defizienz zugrunde. Dieser Enzymdefekt führt zu einer Störung im Purinstoffwechsel und in der weiteren Folge zu einer erhöhten Harnsäureproduktion, die die Ausscheidungsfähigkeit der Nieren überschreitet und teils schwere neurologische Symptome zur Folge hat. Bei heterozygoten Trägerinnen können Metabolite zwischen den mutierten und Wildtyp-Zellen ausgetauscht werden, so dass es bei Frauen nicht zur Ausprägung einer Symptomatik kommt.
Nicht bei jeder X-gebunden rezessiven Erkrankung führt eine zelluläre Interaktion bei heterozygoten Trägerinnen zu einer milderen Symptomatik. Bei der Kranio-fronto-nasalen Dysplasie zeigen Frauen sogar einen schwereren Phänotyp. Es wird durch Mutationen im EFNB1-Gen verursacht, das für ein Protein der Ephrin-Familie codiert. Diese Proteine sind essentielle Bestandteile der Zell-Zell-Kommunikation, Zellsortierung und Zellmigration im Entwicklungsprozess des Embryos. Bei heterozygoten Trägerinnen kommt es durch das Mosaik an Zellen, die das Ephrin B1-Protein exprimieren und defizienten Zellen zu einer gestörten Zellsortierung und Zellmigration. Dieser Effekt wird als zelluläre Interferenz (cellular interference) bezeichnet. Bei hemizygoten Männern besteht eine vollständige Defizienz von Ephrin B1, und man nimmt an, das dessen Funktion von einem anderen Protein der gleichen Familie übernommen wird und sie deshalb keine Symptome zeigen.
Ungleiche / verschobene X-Inaktivierung
In der Regel erfolgt die X-Inaktivierung zufällig und resultiert in einer ca. 50:50 Verteilung von Xmaktiv zu Xpaktiv. Bei verschiedenen X-chromosomalen Erkrankungen konnte jedoch bei Mutationsträgerinnen eine verschobene X-Inaktivierung nachgewiesen werden, die in vielen Fällen einen Einfluss auf den Schweregrad der Erkrankung hat. In einzelnen Studien konnte auch bei zahlreichen gesunden Frauen eine ungleiche X-Inaktivierung beobachtet werden. Verschiedene Faktoren können zu einer ungleichen X-Inaktivierung führen:
- zum Zeitpunkt der X-Inaktivierung besteht der Embryo nur aus wenigen Zellen, so dass aus stochastischen Gründen ungleiche Verhältnisse bei der X-Inaktivierung entstehen können
- genetische Varianten in regulatorischen Bereichen, die die X-Inaktivierung steuern
- negative Selektion: X-gebundene Mutation verursacht bei den Zellen, die diese auf ihrem aktiven X-Chromosom tragen, Schäden, die zur Hemmung des Wachstums oder zum Absterben der Zellen führen.
- positive Selektion (seltener): X-gebundene Mutation verursacht bei den Zellen, die diese auf ihrem aktiven X-Chromosom tragen, einen Wachstumsvorteil
- Trisomie Rettungsmechanismus (trisomy rescue): Zellen, die in der frühen Embryonalentwicklung eine Trisomie aufweisen, zeigen ein schlechtes Wachstum bzw. werden ausselektiert, wodurch der frühe embryonale Zellpool reduziert und eine verschobene X-Inaktivierung begünstigt wird.
Eine verschobene X-Inaktivierung kann in manchen Fällen auch nur in bestimmten Geweben auftreten. Der Selektionsdruck auf Zellen, die eine X-gebundene Mutation tragen, ist nicht in allen Geweben gleich stark, so dass manche Erkrankungen nur eine gewebespezifische ungleiche X-Inaktivierung zeigen. Dies ist zum Beispiel bei der Agammaglobulinämie Bruton, beim X-gebundenen schweren kombinierten Immundefekt (X-SCID) und beim Wiskott Aldrich Syndrom zu beobachten.
Eine weitere Auffälligkeit ist, dass eine ungleiche X-Inaktivierung mit dem Alter korreliert. Studien haben gezeigt, dass bei älteren Frauen häufiger eine ungleiche X-Inaktivierung vorkommt. Daher gibt es X-chromosomale Erkrankungen, die bei Frauen erst später durch die altersbedingte ungleiche X-Inaktivierung auftreten. Hierzu zählen die sideroblastische Anämie und die X-gebundene hämolytische Anämie bedingt durch eine G6PD-Defizienz.
Durch eine verschobene X-Inaktivierung kann es auch bei Frauen zur vollen Ausprägung einer X-gebunden rezessiven Erkrankung kommen, die sich sonst in der Regel nur bei Männern manifestiert. In der Literatur sind Fälle beschrieben bei
- ATRX Syndrom
- Hämophilie B
- Morbus Fabry
- Myotubuläre Myopathie
- Wiskott-Aldrich Syndrom
- X-gebundene hämolytische Anämie
- X-gebundene Thrombozytopenie
Bei manchen X-gebundenen Erkrankungen ist eine Mutation in hemizyogter Form nicht mit dem Leben vereinbar und daher nur bei heterozygoten Trägerinnen zu finden. Durch Mosaike der X-Inaktivierung oder durch eine verschobene X-Inaktivierung kann sich bei betroffenen Frauen eine variable Symptomatik zeigen. Im Falle des Rett-Syndroms, einer schweren neurodegenerativen Erkrankung, zeigt sich bei den Betroffenen in der Regel eine zufällige X-Inaktivierung und daher ein schweres Krankheitsbild. In seltenen Fällen sind Mutationsträgerinnen aufgrund einer stark verschobenen X-Inaktivierung gesund und zeigen keine Symptomatik.
Die X-Inaktivierung kann einen großen Einfluss auf den Phänotyp und den Schweregrad von X-gebundenen Erkrankungen haben und dadurch deren Diagnose erschweren. Die Besonderheiten im Vererbungsmodus und die zahlreichen Faktoren, welche die X-Inaktivierung beeinflussen, spielen bei der humangenetischen Beratung von betroffenen Familien eine wichtige Rolle.
Sun et al. 2022, Genet Res (Camb) 2022:1391807 / Pereira et al. 2021, J Genet 100:63 / Migeon 2020, Genet Med 22:1156 / Migeon 2017, Trends Genet 33:899 / Agrelo et al. 2010, Semin Cell Dev Biol 21:194 / Wutz et al. 2007, Curr Opin Genet Dev 17:387
letzte Aktualisierung: 1.11.2023