Tuberöse Sklerose Complex (TSC)
Die Tuberöse Sklerose (TSC) ist eine autosomal-dominant vererbte Multisystemerkrankung mit variabler klinischer Symptomatik. Ursächlich sind pathogene Varianten in den TSC1- und TSC2-Genen, die als Tumorsuppressorgene fungieren. Diese Gene wirken auf zellulärer Ebene rezessiv, d.h., sie führen nur dann zur lokalen Entstehung von Hamartomen, wenn durch zwei unabhängige Varianten beide homologen TSC-Gene inaktiviert wurden. Infolge von pathogenen TSC1- oder TSC2-Varianten kommt es zur Überaktivierung der mTOR-Signaltransduktion und zu einer verstärkten Proliferation in den charakteristischen TSC-Läsionen.
Wissenschaftlicher Hintergrund
Die Tuberöse Sklerose (TSC, Tuberous Sclerosis Complex, M. Bourneville-Pringle) ist eine autosomal-dominant vererbte Multisystemerkrankung mit großer klinischer Variabilität. Die Inzidenz wird mit ca. 1:7.000 angegeben. Charakteristisch sind multiple, lokale Areale unvollständiger und abnormer Gewebedifferenzierung, sog. Hamartien, die bei verstärkter Proliferation als Hamartome bezeichnet werden, aber gutartig bleiben. TSC kann sich in fast allen Organen manifestieren, wobei Gehirn, Herz, Nieren, Lunge, Haut und Augen am häufigsten betroffen sind. Die Organmanifestationen sind jedoch alle fakultativ, keines dieser Symptome ist immer nachweisbar. Einige Symptome haben keinen Krankheitswert, weisen jedoch darauf hin, dass die betroffene Person Anlageträger ist. Anhand der aktualisierten, 2013 veröffentlichten diagnostischen Kriterien kann die Diagnose Tuberöse Sklerose (TSC) sowohl genetisch als auch klinisch gestellt werden. Demnach ist der alleinige Nachweis einer pathogenen Variante im TSC1– oder TSC2-Gen ausreichend für die Diagnosestellung. Die klinischen Manifestationen werden in 11 Haupt- und 6 Nebenkriterien eingeordnet, wobei sowohl zwei Hauptkriterien als auch die Kombination von einem Haupt- und mindestens zwei Nebenkriterien TSC sichern, während entweder ein Haupt- oder mindestens zwei Nebenkriterien TSC möglich machen.
Molekulare Ursachen sind pathogene Varianten im TSC1– und TSC2-Gen. In Familien mit mehreren Betroffenen sind Varianten des TSC1– und des TSC2-Gens gleich häufig, 70% der TSC-Fälle treten allerdings sporadisch durch Neumutationen auf, wobei in diesen Fällen nur in 10-15% TSC1 und in 70% TSC2 verändert ist. Insgesamt sind TSC2-Varianten drei- bis viermal häufiger als TSC1-Varianten. Bei beiden TSC-Genen handelt es sich um Tumorsuppressor-Gene, die auf zellulärer Ebene rezessiv wirken, d.h. nur dann zur lokalen Entstehung von Hamartomen führen, wenn durch zwei unabhängige Varianten beide homologen TSC-Gene inaktiviert wurden. Die TSC1– und TSC2-Genprodukte Hamartin und Tuberin bilden einen Komplex und haben eine zentrale Funktion innerhalb grundlegender Signaltransduktionswege, über die Zelladhäsion, Transkription und Zellproliferation, Vesikeltransport und Zellmigration gesteuert werden. Eine zentrale Rolle stellt die Insulin-vermittelte mTOR-Signaltransduktion dar. Der Tuberin-Hamartin-Komplex inhibiert die Aktivität der Serin-Kinase mTOR (mammalian Target of Rapamycin). Infolge von pathogenen TSC1– oder TSC2-Varianten kommt es zur Überaktivierung der mTOR-Signaltransduktion und zu einer verstärkten Proliferation in den charakteristischen TSC-Läsionen. Durch die Interaktion von Hamartin und Tuberin führt die Inaktivierung beider Kopien eines der beiden TSC-Gene zum Funktionsverlust des gesamten Proteinkomplexes und somit zur gleichen Pathogenese. Bis auf einige Ausnahmen ist keine klare Genotyp-Phänotyp-Korrelation möglich.
Bei Ausfall des TSC1/TSC2-Komplexes kann die aktivierte mTOR-Signaltransduktion auch durch Medikamente gehemmt werden. Die in der Therapie Bei TSC-Patienten eingesetzten mTOR-Inhibitoren Rapamycin (Sirolimus, Rapamune®) und dessen Derivat RAD001 (Everolimus, Votubia®) hemmen das Wachstum von Angiomyolipomen der Niere und von Riesenzellastrozytomen des Gehirns und können darüber hinaus die Angiofibrome im Gesicht, die Epilepsie und die respiratorische Funktion bei Lymphangiomyomatose positiv beeinflussen.
Die Varianten sind in beiden TSC-Genen über nahezu alle Exons bzw. angrenzende Intronsequenzen verteilt und umfassen alle Mutationstypen. Im TSC1-Gen machen Varianten, die zum vorzeitigen translationalen Stop führen, mit ca. 90% den Hauptanteil aus. Dagegen sind pathogene Missense-Varianten und größere genomische Deletionen mit weniger als 6% bzw. 3% relativ selten. Im TSC2-Gen sind alle Arten von kleinen Nukleotidveränderungen etwa gleich häufig, wobei die Konsequenz ebenfalls in 75% ein vorzeitiger translationaler Stop ist. Verluste größerer Genbereiche machen etwa 5% aus, wobei in 4,5% Teile des Gens und 0,5% das gesamte Gen betreffen. Von den kompletten Gendeletionen ist in der Hälfte neben dem TSC2-Gen zusätzlich das chromosomal benachbarte PKD1-Gen für die autosomal-dominante polyzystische Nierenerkrankung (ADPKD) betroffen. Patienten mit diesem TSC2/PKD1 Contiguous Gene Syndrome weisen sowohl klinische Merkmale einer TSC als auch einer ADPKD mit früher Manifestation von Nierenzysten auf.
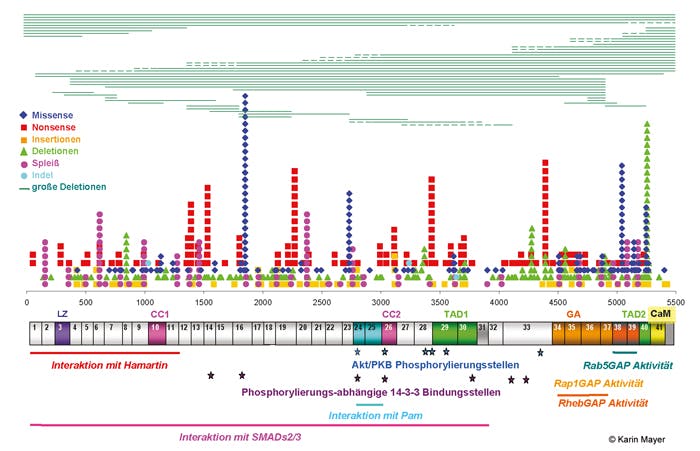
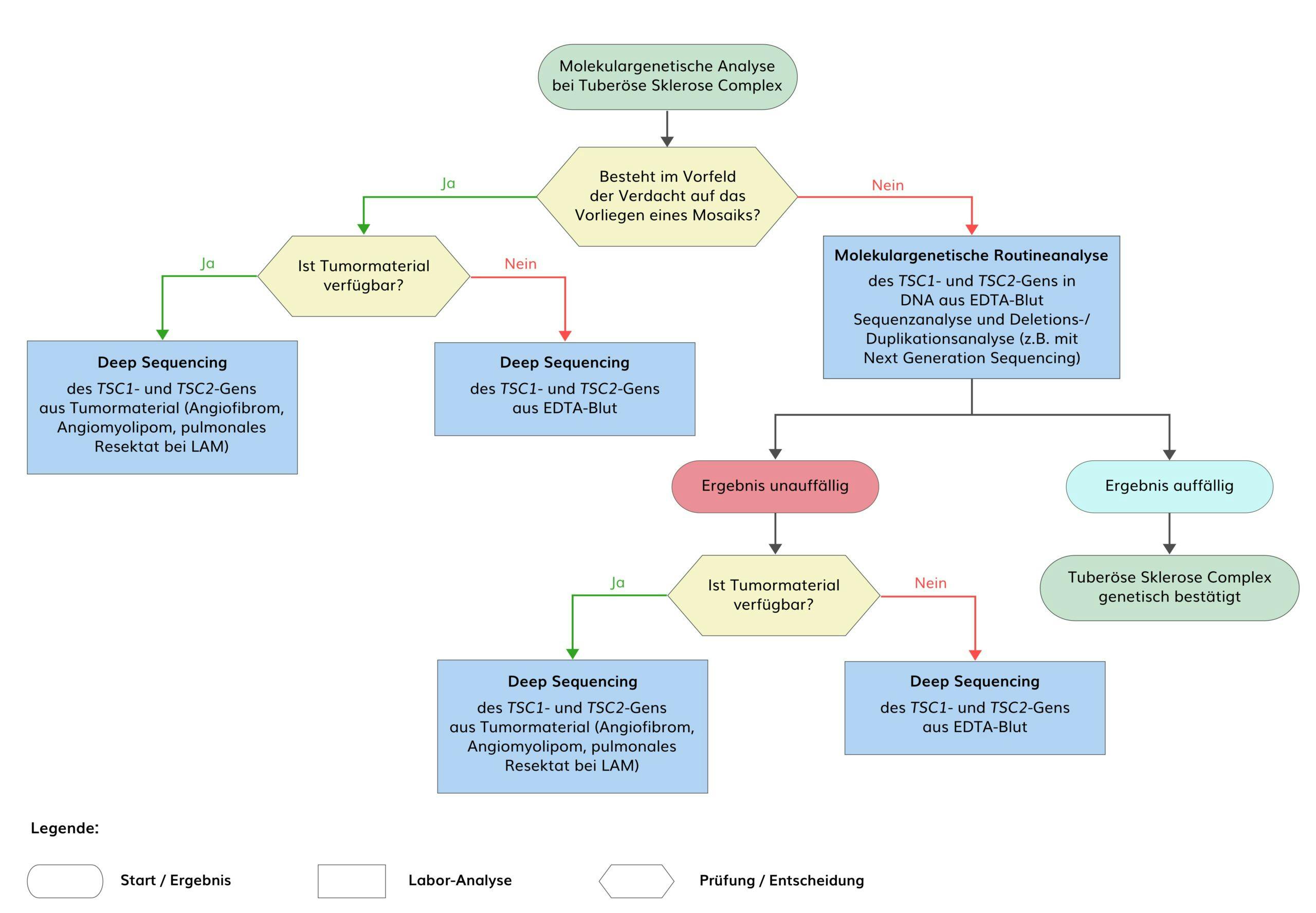
Die genetische Analyse beider TSC-Gene mittels Sequenzierung erfasst ca. 90% aller bisher bekannten Nukleotidveränderungen. Bis zu 10% der pathogenen Varianten in beiden TSC-Genen sind größere genomische Deletionen und Duplikationen. Bei etwa 85% aller Patienten mit der klinisch gesicherten Diagnose TSC kann durch die Kombination von Sequenzanalyse der codierenden Regionen und zusätzlicher Deletions-/Duplikationsdiagnostik eine pathogene Variante in einem der beiden TSC-Gene detektiert werden. Bei etwa 50% der mit dieser Methodik molekulargenetisch nicht aufzuklärenden Fälle können mittlerweile pathogene Varianten in Form von Mosaiken oder in regulatorischen Regionen mit Hilfe von Next Generation Sequencing mit einer Sequenziertiefe von mind. 1000-fach (NGS; Deep Sequencing) der gesamten genomischen Regionen beider TSC-Gene detektiert werden.
Nachweis von Mosaiken bei Tuberöse Sklerose Complex (TSC)
Bei etwa 15% aller Patienten mit der klinisch gesicherten Diagnose Tuberöse Sklerose Complex (TSC) kann durch die Kombination von Sequenzanalyse der codierenden Regionen inkl. der flankierenden Bereiche (+/- 5 Nukleotide) mit Sanger-Sequenzierung oder mit Next Generation Sequencing (NGS) mit einer Sequenziertiefe von 30-fach und zusätzlicher und Deletions-/Duplikationsdiagnostik mit MLPA oder mittels Gendosisanalyse aus NGS-Daten keine pathogene Variante in einem der beiden TSC-Gene detektiert werden. Bei einem Teil dieser Patienten liegen genetische Mosaike vor, wobei der Anteil der Zellen mit Varianten im TSC1– oder TSC2-Gen im untersuchten Gewebe unter der Nachweisgrenze der Sanger-Sequenzierung liegt. Ein weiterer Anteil von Varianten befindet sich in den regulatorischen Regionen beider TSC-Gene. Durch die Analyse der gesamten genomischen Regionen beider TSC-Gene mit einer Sequenziertiefe (coverage) von mindestens 1.000x (Deep Sequencing) ist es möglich, Mosaike mit einem Anteil der Variante (VAF) < 5% und Intronvarianten jenseits der Exon/Intron-Übergänge zu detektieren.
Die Prävalenz von post-zygotischen Mosaiken bei TSC, bei denen eine Variante im TSC1– oder TSC2-Gen während der Embryogenese entstanden ist, wird auf 7,5-15% geschätzt, wobei große genomische Deletionen im Mosaik bisher leichter zu detektieren waren als Einzelnukleotidveränderungen. Bei Patienten mit TSC-Varianten im Mosaik sind meistens weniger Organsysteme betroffen als bei Patienten mit heterozygoten Varianten. Die Organbeteiligung korreliert mit dem Anteil des mutierten Allels in DNA in dem Gewebe, in dem die Inaktivierung des zweiten Allels zur Entstehung eines Hamartoms geführt hat, wobei dieser außer in DNA aus betroffenen Hautläsionen oft schwer zu bestimmen ist. Da bei Mosaiken der Anteil des mutierten Allels in DNA aus Leukozyten in vielen Fällen auch unter der Detektionsgrenze von NGS mit einer coverage von 1000x liegt, empfiehlt sich in diesen Fällen eine Analyse mittels Deep Sequencing aus Tumorgewebe, z.B. Hautläsionen.
HH, Pagon RA, et al, eds. GeneReviews® (Updated 2018 July 12) / Martin et al. 2017, Nat Commun 8:15816 / Northrup and Krueger 2013, Pediatr Neurol 49:243 / Mayer et al. 2013, Eur J Hum Genet Jun 12. doi: 10.1038/ejhg.2013.129 / Staley et al. 2011, Pediatrics 127:e117 / Kwiatkowski 2010, in Tuberous Sclerosis Complex. Genes, Clinical Features and Therapeutics (Kwiatkowski/Holets Whittemore/Thiele, eds.) chap. II.4: 29-57 / Orlova and Curatolo 2010, Ann N Y Acad Sci 1184:87 / Curatolo et al. 2008, The Lancet 372:657 / Au et al, Genet Med 9:88 (2008) / Kozlowski et al. 2007, Hum Genet 121 :389 / Yates 2006, Eur J Hum Genet 14:1065 / Sancak et al. 2005, Eur J Hum Genet 13:731 / Dabora et al. 2001, Am J Hum Genet 68:64 / Mayer et al. 1999, Hum Mutat 14:401 / Roach 1998, J Child Neurol 13:624
Treichel et al. 2019, Genet Med May 22, Epub ahead of print / Giannikou et al. 2019, Genet Med Jun 4, Epub ahead of print / Mayer et al. 2017, ESHG 2017: Abstract P14.091C / Tyburczy et al. 2015, PLoS Genet 11(11):e1005637 / Nellist et al. 2015, BMC Medical Genetics 16:10 / Curatolo et al. 2015, Semin Pediatr Neurol 22:259 / Boronat et al. 2014, Clin Genet 86:149 / Gutte et al. 2013, Indian J Dermatol 58:159 / Alshaiji et al. 2012, Dermatol Online J 18:8 / Mayer et al. 2012, Eur J Hum Genet 20 (Supp1):287, P11.132 / Qin et al. 2010, Hum Genet 127:573 / Mayer et al. 2000, Biochim Biophys Acta 1502:495
letzte Aktualisierung: 19.3.2024