Neurofibromatose Typ 1 (NF1)
Die Neurofibromatose Typ 1 (NF1), auch bekannt als M. Recklinghausen, ist eine der häufigsten erblichen neurologischen Erkrankungen. Sie wird autosomal-dominant vererbt, in 50% der Erkrankungen ensteht sie jedoch durch Neumutationen. Charakteristische Merkmale sind kutane und subkutane Neurofibrome, Café-au-lait-Flecken der Haut, sommersprossenartige Flecken in der Achselhöhle oder in der Leiste und Lisch-Knötchen der Iris. Ursächlich sind pathogene loss-of-function Varianten im NF1-Gen. In mindestens 5% der Patienten liegt eine Mikrodeletion im Chromosom 17q11.2 vor, die das NF1-Gen beinhaltet. Die Funktion des NF1-Genprodukts Neurofibromin ist die eines Tumorsuppressors, indem durch die Aktivierung einer Ras-GTPase die zelluläre Proliferation kontrolliert wird.
Wissenschaftlicher Hintergrund
Die Neurofibromatose Typ 1 (NF1, M. Recklinghausen) gehört mit einer Inzidenz von 1:3.000 zu den häufigstenerblichen neurologischen Erkrankungen, die mit gutartigen und bösartigen Tumoren des Nervensystems assoziiert ist. Charakteristisch sind kutane und subkutane Neurofibrome, die typischerweise in der Adoleszenz auftreten, Café-au-lait-Flecken der Haut, die sich meist in der ersten Lebensdekade manifestieren, sommersprossenartige Flecken in der Achselhöhle oder in der Leiste und Lisch-Knötchen der Iris. Seltener treten schwerwiegende klinische Manifestationen wie plexifome Neurofibrome, Optikusgliome, Neurofibrosarkome und Knochenveränderungen auf. Die diagnostischen Kriterien für NF1 wurden 1988 von den National Institutes of Health (NIH) erarbeitet. NF1 zeigt eine vollständige Penetranz bei hoher phänotypischer Variabilität. Die Vererbung ist autosomal-dominant, bei 50% der Patienten besteht eine positive Familienenamnese, während 50% der Erkrankungen durch Neumutationen entstehen.
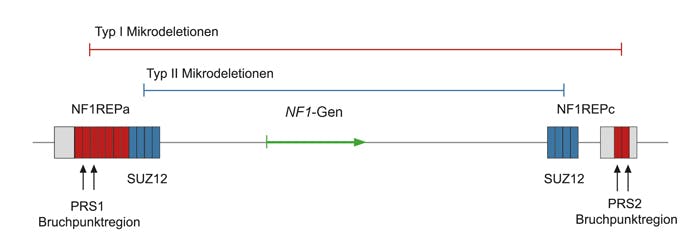
Genetische Ursache für NF1 sind pathogene loss-of-function Varianten im NF1-Gen. Die Varianten sind über nahezu alle Exons bzw. angrenzende Intronsequenzen verteilt und umfassen alle Mutationstypen, wobei translationale Stopmutationen mit 80% am häufigsten vorkommen. 22-30% der Nukleotidveränderungen beeinflussen den Spleißvorgang, 1/3 davon sind alleine mit DNA-basierten Methoden nicht nachzuweisen. In mindestens 5% der Patienten liegt eine Mikrodeletion Typ 1 (1,4 Mb), Typ 2 (1,2 Mb), Typ 3 (1,0 Mb) oder eine atypische Mikrodeletion im Chromosom 17q11.2 vor, die das NF1-Gen beinhaltet. Diese Mikrodeletionen sowie intragene Deletionen, die ein oder mehrere Exons betreffen, machen etwa 2% aller Varianten im NF1-Gen aus. Duplikationen des gesamten NF1-Gens und der flankierenden genomischen Region führen im Gegensatz zu Mikrodeletionen nicht zu einem charakteristischen NF1-Phänotyp. Die betroffenen Patienten können eine Entwicklungsverzögerung und eine Epilepsie aufweisen. Die Mutationsrate im NF1-Gen ist mit 1:10.000 eine der höchsten im menschlichen Genom. Bei der Hälfte der Patienten liegt eine Neumutation vor. Die Funktion des NF1-Genprodukts Neurofibromin ist die eines Tumorsuppressors, indem durch die Aktivierung einer Ras-GTPase die zelluläre Proliferation kontrolliert wird.
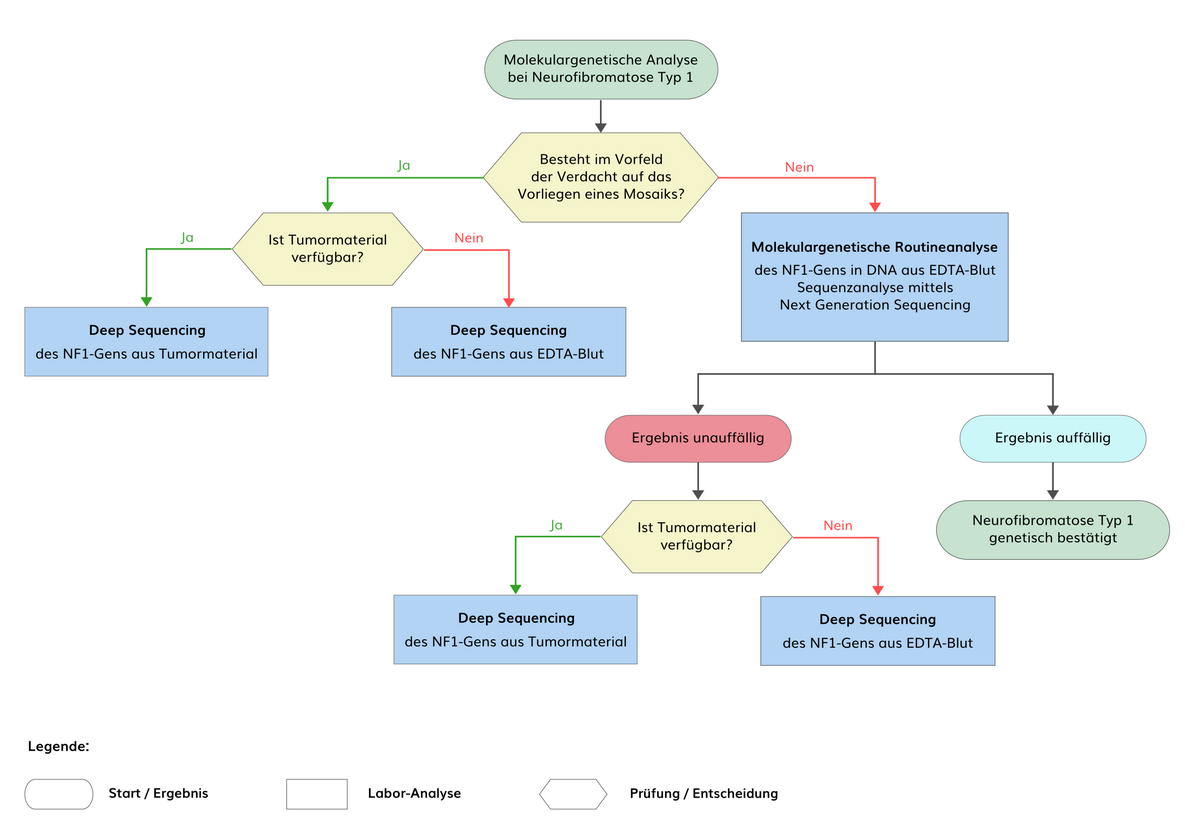
Mit einer Kombination mehrerer Analysetechniken, die eine Sequenzanalyse der codierenden Regionen, eine Deletions-/Duplikationsanalyse und eine cDNA-Analyse beinhalten, können bei mehr als 95% der Patienten, bei denen die diagnostischen Kriterien des NIH Consensus Development Conference Statement für Neurofibromatose Typ 1 (NF1) erfüllt sind, pathogene Varianten im NF1-Gen nachgewiesen werden. Insbesondere cDNA-Analysen sind allerdings nicht Bestandteil der Routinediagnostik.
Differentialdiagnostisch zu NF1 ist das Legius-Syndrom zu nennen. Bei Patienten mit Legius-Syndrom sind die diagnostischen NIH-Kriterien ebenfalls erfüllt. Ein charakteristischer Unterschied zu NF1 ist das Fehlen von Lisch-Knötchen der Iris und Neurofibromen und im Gegensatz dazu das Auftreten von subkutanen Lipomen im Erwachsenenalter. Molekulare Ursache des Legius-Syndroms sind heterozygote Varianten im SPRED1-Gen.
Eine weitere Differenzialdiagnose in Bezug auf die Hautmanifestationen stellt das konstitutionelle Mismatch-Repair-Defizienz-Syndrom (CMMRDS) dar. CMMRDS ist ein seltenes, kindliches Tumorprädispositionssyndrom mit klinisch überlappenden Symptomen zu NF1 wie Cafe-au-lait Flecken, Freckling, und Lisch-Knötchen. Die Erkrankung ist charakterisiert durch das Auftreten verschiedener Tumore wie z.B. Hirntumore, Tumore des Gastrointestinaltrakts, hämatologischen Neoplasien oder auch seltenerer Tumore (z.B. Sarkome). Molekulare Ursache des autsomal-rezessiv vererbten CMMRDS sind homozygote oder kombiniert heterozygote pathogene Varianten in einem der vier Mismatch-Repair-Gene MLH1, MSH2, MSH6 oderPMS2. Ein Hinweis auf CMMRDS ist das Vorliegen von klinischen Symptomen eines Lynch-Syndroms bei einem oder beiden Elternteilen.
Nachweis von Mosaiken bei Neurofibromatose Typ 1 (NF1)
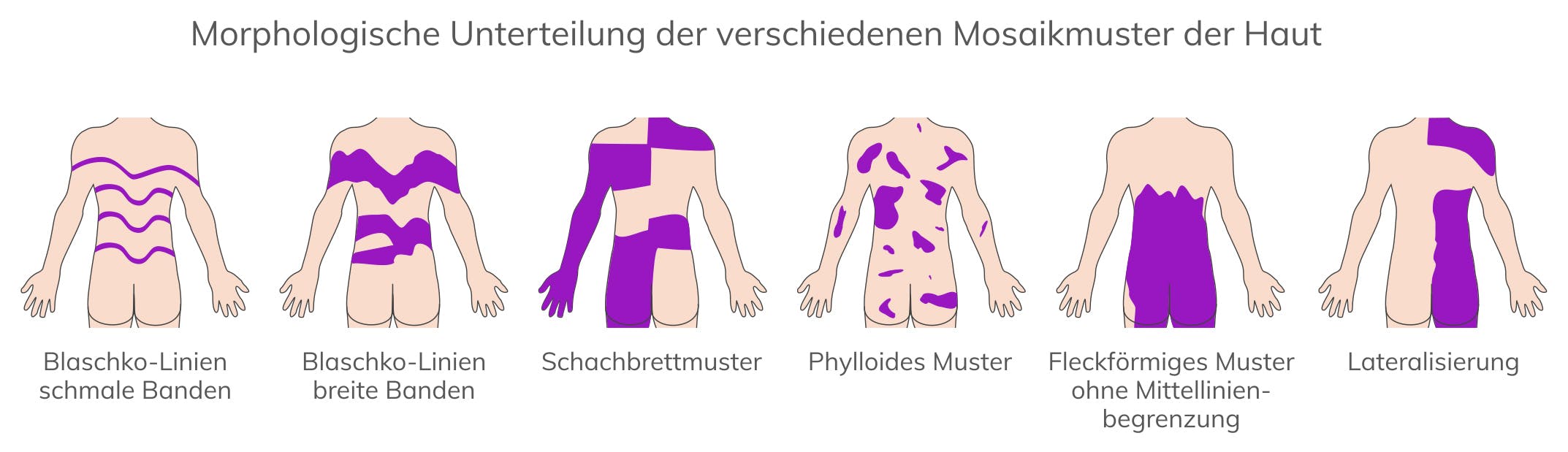
Bis zu 10% aller pathogenen Varianten im NF1-Gen liegen in Form von somatischen Mosaiken vor. Der höchste Anteil an Mosaiken kommt bei Typ 2-Mikrodeletionen und atypischen Mikrodeletionen vor.
Mosaik-Neurofibromatose Typ 1 (MNF) entsteht durch eine postzygotische genetische Veränderung während der Embryonalentwicklung. Je nach Zeitpunkt des Mutationsereignisses und betroffener Zellpopulation können sich isolierte Pigmentierungsstörungen (Café-au-lait-Flecken mit oder ohne Freckling), Neurofibrome oder plexiforme Neurofibrome entwickeln, die häufig jeweils unilateral auftreten. Sofern die NF1-charakteristischen Manifestationen auf spezifische Körperregionen beschränkt sind, wird dies auch als segmentale NF1 bezeichnet. Die Prävalenz der Mosaik-Neurofibromatose wird auf 1:36.000 geschätzt. Bei Mosaik-Neurofibromatose Typ 1 ist in DNA aus peripheren Leukozyten eine genetische Veränderung im NF1-Gen nicht oder nur in geringen Anteilen <5% nachweisbar. Bei isolierten Pigmentierungsstörungen kann eine genetische Veränderung in kultivierten Melanozyten aus der betroffenen Region nachgewiesen werden. Bei isolierten Neurofibromen kann eine genetische Veränderung nur in Schwann-Zellen aus dem entsprechenden Tumor und ggf. in der darüber liegenden Haut nachgewiesen werden.
Pathogene Varianten in den Introns und regulatorische Varianten machen 1-3% aller NF1-Veränderungen aus. Varianten in den nicht-codierenden Bereichen könnten mit Hilfe von NGS der gesamtgenomischen Region des NF1-Gens (Deep Sequencing) und mit Hilfe von RNA-Analysen nachgewiesen werden, wobei diese Analysen derzeit nicht Bestandteil der Routinediagnostik sind.
Erkrankung | ICD—10 | Gen | OMIM—G |
| Neurofibromatose Typ 1 | Q85.0 | NF1 | 613113 |
Friedman In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews® (Last Revision: June 6, 2019) / Kehrer-Sawatzki et al. 2017, Hum Genet 136:349 / Wimmer et al. 2017, Clin Genet 91:507 / Evans et al. 2016, EBioMedicine 7:212 / Pasmant et al. 2015, Eur J Hum Genet 23:596 / van Minkelen et al. 2014, Clin Genet 85:318 / Sabbagh et al. 2013, Hum Mutat 34:1510 / Vogt et al. 2012, Hum Mutat 33:1599 / Valero et al. 2011, J Mol Diagn 13:113 / Messiaen and Wimmer 2008, In: Kaufmann D, ed. Monographs in Human Genetics. Vol 16:63 / NIH Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987, Neurofibromatosis 1:172
Lara-Corrales et al. 2017, J Cutan Med Surg 21:379 / Kehrer-Sawatzki et al. 2017, Hum Genet 136:349 / García-Romero et al. 2016, Pediatr Dermatol 33:9 / Evans et al. 2016, EBioMedicine 7:212 / Soares Cunha et al. 2016, Genes 7:133 / Svaasand et al. 2015, Hered Genet Curr Res 4:3 / Sabbagh et al. 2013, Hum. Mutat 34:1510 / Garcia-Linares et al. 2011, Hum Mutat 32:78; Messiaen et al. 2011, Hum Mutat 32:213 / De Schepper et al. 2008, J Invest Dermatol 128:1050 / Maertens et al. 2007, Am J Hum Genet 81:243 / Consoli et al. 2005, J Invest Dermatol 125:463
letzte Aktualisierung: 19.3.2024