Lynch-Syndrom
Lynch-Syndrom, auch bekannt als hereditäres nicht-polypöses Kolonkarzinom (HNPCC), ist für etwa 2-3% aller Fälle von kolorektalem Karzinom verantwortlich und zeigt oft ein junges Erkrankungsalter sowie multiple Tumoren. Die Diagnose basiert auf klinischen Kriterien wie den Amsterdam-II- und Bethesda-Kriterien. Die Erkrankung wird durch pathogene Varianten in den Mismatch-Repair-Genen (MMR-Gene) verursacht, wobei MLH1 und MSH2 am häufigsten betroffen sind. Eine stufenweise Diagnostik, die immunhistochemische und molekularpathologische Untersuchungen umfasst, ist entscheidend für die Identifikation des Syndroms und die prädiktive Diagnostik bei Verwandten.
Wissenschaftlicher Hintergrund
Das kolorektale Karzinom (CRC) gehört zu den häufigsten Tumorerkrankungen der westlichen Industrienationen. Bei etwa 10% der Fälle ist eine familiäre Häufung zu beobachten. Etwa 2-3% aller CRC-Erkrankungen sind auf das Lynch-Syndrom bzw. hereditäre nicht-polypöse Kolonkarzinom (HNPCC) Syndrom zurückzuführen. Patienten mit Lynch-Syndrom weisen häufig ein auffällig junges Erkrankungsalter auf und/oder erkranken an multiplen Tumoren (vorallem gastrointestinale, gynäkologische Tumoren, Tumoren des Urogenitaltrakts und der Gallenwege). Das Erkrankungsrisiko kann bis zu 80-90% betragen. Eine seltene phänotypische Variante des HNPCC ist das Muir-Torre-Syndrom, bei dem zusätzlich Talgdrüsentumoren und Keratoakanthome der Haut beobachtet werden.
Die Diagnose HNPCC wird klinisch gestellt, wenn die sogenannten Amsterdam-II-Kriterien erfüllt sind (alle Kriterien müssen erfüllt sein):
- mindestens 3 Familienangehörige mit histologisch gesichertem kolorektalen Karzinom (oder einem Karzinom des Endometriums, Dünndarms, Ureters oder Nierenbecken), einer davon mit den beiden anderen erstgradig verwandt;
- Erkrankung in mindestens zwei aufeinanderfolgenden Generationen;
- mindestens ein Patient mit Diagnosestellung vor dem 50. Lebensjahr; und
- vorangegangener Ausschluss einer Familiären adenomatösen Polyposis (FAP).
Aufgrund der abnehmenden Größe von Familien sowie zum Teil unvollständiger Penetranz werden die Amsterdam-II-Kriterien jedoch nur noch selten erfüllt. Daher wurden die revidierten Bethesda-Kriterien formuliert, um weitere potentielle HNPCC-Patienten zu identifizieren (mindestens ein Kriterium muss erfüllt sein):
- Patient mit CRC vor dem 50. Lebensjahr;
- Patient mit synchronen oder metachronen CRC oder anderen HNPCC-assoziierten Tumorerkrankungen [Endometrium, Magen, Ovarien, Pankreas, Urothel, Gallengänge, Dünndarm, Gehirn, Talgdrüsenadenome, Keratokanthome (Muir-Torre-Syndrom)], unabhängig vom Alter;
- Patient unter 60 Jahren mit CRC mit MSI-Histologie (lymphozytäre Infiltration, muzinöse und/oder Siegelzellring-Differenzierung bzw. medullärem Wachstum);
- Patient mit CRC (unabhängig vom Alter) und einem erstgradig Verwandten mit CRC oder einem HNPCC-assoziierten Tumor vor dem 50. Lebensjahr; oder
- Patient mit CRC (unabhängig vom Alter) und mind. zwei erst- oder zweitgradig Verwandten mit einem CRC oder einem HNPCC-assoziierten Tumor (unabhängig vom Alter).
Tumoren von Patienten, die die revidierten Bethesda-Kriterien erfüllen, sollten auf eine Mismatch-Reparatur-Defizienz hin untersucht werden (MSI-Analyse und Immunhistochemie an Tumormaterial, vgl. Stufendiagnostik).
Ursächlich für das Lynch-Syndrom sind pathogene Varianten in den Mismatch-Repair-Genen (MMR-Gene) MLH1, MSH2, MSH6 oder PMS2. Seltener können auch Deletionen von Teilen des EPCAM-Gens die Expression von MSH2 mindern und so ein Lynch-Syndrom verursachen.
Das Vorliegen einer pathogenen Variante in einem der MMR-Gene führt nicht unmittelbar zur Tumorentstehung, erst nach Ausfall des zweiten, intakten Allels durch spontane somatische Ereignisse kommt es zum Funktionsverlust des Gens und möglicherweise zur Entartung der betroffenen Zelle (Zwei-Treffer-Hypothese nach Knudson). In den meisten Fällen sind ursächliche Varianten in MLH1 oder MSH2 nachweisbar (40% bzw. 34%), MSH6, PMS2 oder EPCAM sind deutlich seltener betroffen (18%, 8%, variabel). Das Risiko für die Entstehung einer Tumorerkrankung ist abhängig vom betroffenen Gen: MSH2-Varianten sind mit einem 57-80%-igen Risiko assoziiert, bei Vorliegen von MLH1-Varianten beträgt es 59-65%, MSH6-Anlageträger besitzen ein etwa 25%-iges (Männer) bzw. 40%-iges Risiko (Frauen), PMS2-Varianten sind mit einem Risiko von etwa 25-32% assoziiert
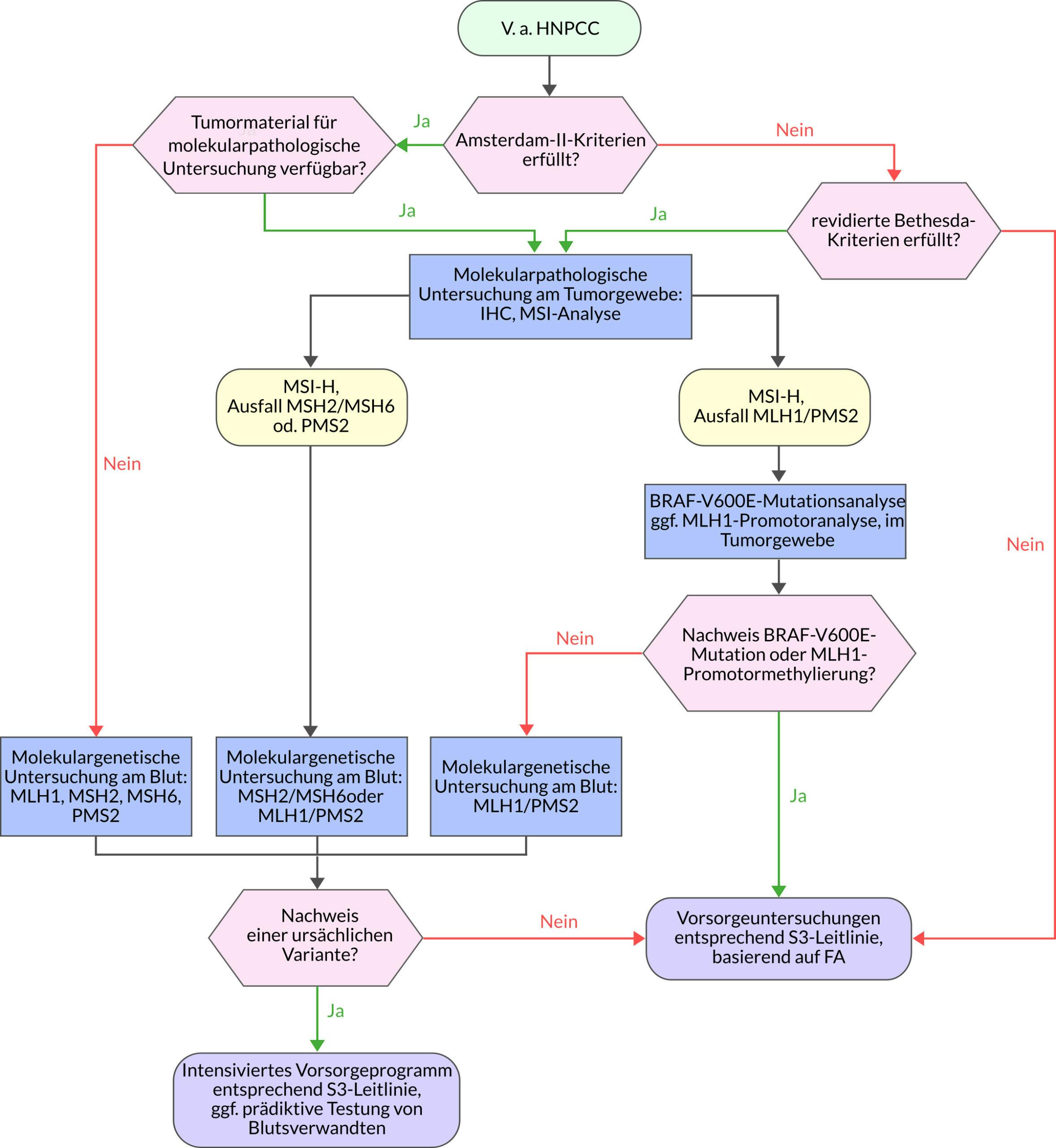
Stufendiagnostik
Die HNPCC-Diagnostik erfolgt stufenweise (vgl. interaktives Flussdiagramm zur HNPCC-Diagnostik):zunächst sollte eine erkrankte Person (Indexpatient) untersucht werden. Anfangs wird Tumorgewebes des Indexpatienten immunhistochemisch auf einen Ausfall eines der MMR-Proteine bzw. molekularpathologisch auf Vorliegen einer Mikrosatelliteninstabilität (MSI) untersucht. Bei Nachweis eines MSH2/MSH6-Ausfalls kann die Analyse dieser Gene in der Keimbahn erfolgen. Bei Expressionsverlust von MLH1/PMS2 kann durch die BRAF-V600E Analyse und ggf. MLH1-Promotoranalyse am Tumor weiter zwischen einem sporadischen Tumor oder einem Tumor im Rahmen eines Lynch-Syndroms differenziert werden. Bei unauffälligem BRAF– und MLH1-Promotor-Befund kann die Keimbahndiagnostik von MLH1/PMS2 angeschlossen werden.
Die direkte molekulargenetische Untersuchung aller vier MMR-Gene bei gesetzlich Versicherten kann derzeit nur durchgeführt werden, wenn eine vorausgehende molekularpathologische Untersuchung am Tumormaterial nicht möglich ist und klinisch ein Lynch-Syndrom gesichert ist (Erfüllung aller Amsterdam-II-Kriterien).
Wird beim Indexpatienten eine krankheitsverursachende Keimbahnvariante in einem der vier MMR-Gene nachgewiesen, können gesunde Blutsverwandte gezielt auf diese Variante untersucht werden (prädiktive Diagnostik). Bei einer prädiktiven Diagnostik muss entpsrechend GenDG vor der Untersuchung und nach Vorliegen des Resultats genetisch beraten werden (§10, Abs. 2 GenDG).
Entsprechend der S3-Leitlinie Kolorektales Karzinom sollten Anlageträger regelmäßige und intensivierte Vorsorgeuntersuchungen im Hinblick auf kolorektale Karzinome und Karzinome im oberen Verdauungstrakt wahrnehmen. Weibliche Anlageträgerinnen sollten zusätzlich regelmäßig auf Endometrium- und Ovarialkarzinome untersucht werden. Außerdem kann eine Hysterektomie und ggf. eine Adnexexstirpation diskutiert werden.
Leitlinienprogramm Onkologie: S3-Leitlinie Kolorektales Karzinom, Langversion 2.1, 2019 / Kohlmannet al. 2018, Lynch Syndrome, GeneReviews®, www.ncbi.nlm.nih.gov/books/NBK1211/ / Leitlinienprogramm Onkologie: Diagnostik, Therapie und Nachsorge der Patientinnen mit Endometriumkarzinom, Langversion 1.0, 2018 / Da Silva et al. 2016, Anticancer Res 36:4399 / Peltomäki 2016, Fam Cancer 15:385 / Shai et al. 2014, Fam Cancer 13:499 / Steinke et al. 2013, Dtsch Arztebl Int 110:32 / Holinski-Feder und Grabowski 2006, medgen 18:246
letzte Aktualisierung: 11.11.2023