Methylentetrahydrofolatreduktase- (MTHFR-) Defizienz
Die Methylentetrahydrofolat-Reduktase- (MTHFR-) Defizienz ist eine seltene, autosomal-rezessiv vererbte Störung des Folsäurestoffwechsels, die zu variablen neurologischen Symptomen führen kann. Durch einen Enzymmangel wird vermindert 5-Methyl-THF für die Remethylierung von Homocystein zu Methionin zur Verfügung gestellt. Die Folgen sind Homocystinurie, Hyperhomocysteinämie und reduzierte Methionin-Plasmakonzentrationen. Deutlich häufiger tritt eine milde Form der MTHFR-Defizienz auf, die durch eine thermolabile Variante des Enzyms mit eingeschränkter Funktion verursacht wird. Molekulargenetisch kann zumeist die häufige C677T-Variante (rs1801133) in Exon 5 des MTHFR-Gens nachgewiesen werden. Eine ausreichende Versorgung mit Folsäure, Vitamin B6 und B12 ist für Träger des T/T-Genotyps empfehlenswert.
Wissenschaftlicher Hintergrund
Die Methylentetrahydrofolat-Reduktase- (MTHFR-) Defizienz ist eine seltene, autosomal-rezessiv vererbte Störung des Folsäurestoffwechsels, die zu variablen neurologischen Symptomen wie Muskelschwäche, Parästhesien, mangelnde Koordination und Einschränkung der Merkfähigkeit führen kann. Das Enzym Methylentetrahydrofolatreduktase (MTHFR) spielt eine wesentliche Rolle im Homocysteinstoffwechsel. MTHFR katalysiert die Reduktion von 5,10-Methylentetrahydrofolat zu 5-Methyl-Tetrahydrofolat (THF), welches ein wichtiger Methylgruppendonor ist.
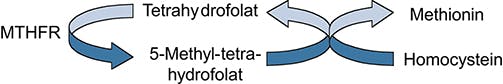
Durch einen Enzymmangel wird vermindert 5-Methyl-THF für die Remethylierung von Homocystein zu der essentiellen Aminosäure Methionin zur Verfügung gestellt. Die Restaktivität der MTHFR ist in der Regel <10%. Die Folgen sind Homocystinurie, Hyperhomocysteinämie (Homocysteinspiegel >100µmol/l) und reduzierte Methionin-Plasmakonzentrationen. Ursächlich sind pathogene Varianten im MTHFR-Gen, das auf dem langen Arm von Chromosom 1 lokalisiert ist.
Deutlich häufiger tritt eine milde Form der MTHFR-Defizienz auf, die durch eine thermolabile Variante des Enzyms mit eingeschränkter Funktion verursacht wird. Diese milde Form ist gekennzeichnet durch erhöhte Homocysteinspiegel (Hyperhomocysteinämie), im Gegensatz zur klassischen Form treten allerdings keine neurologischen Symptome auf. Molekulargenetisch kann zumeist die häufige C677T-Variante (rs1801133) in Exon 5 des MTHFR-Gens nachgewiesen werden. Homozygotie für diese Variante ist mit einem erhöhten Homocysteinspiegel assoziiert. Eine schwach positive Assoziation mit multifaktoriellen Erkrankungen wie z.B. Thrombophilie oder Neuralrohrdefekte (Spina bifida) ist ebenfalls nur für homozygote Merkmalsträger und nur in Kombinationmit weiteren Risikofaktoren beschrieben. Eine ausreichende Versorgung mit Folsäure, Vitamin B6 und B12 ist für Träger des T/T-Genotyps empfehlenswert. Eine weitere Variante im MTHFR-Gen, A1298C (rs1801131), ist in kombinierter Heterozygotie mit der C677T-Variante ebenfalls mit verminderter Enzymaktivität und erhöhten Homocysteinkonzentrationen im Blut assoziiert. Homozygotie für den C/C-Genotyp hat jedoch keine Auswirkung auf den Folat-abhängigen Homocysteinmetabolismus.
Die Bestimmung des MTHFR-Genotyps wird derzeit nur für Patienten mit einer Plasma-Homocysteinkonzentration (tHyc) von >50µmol/L empfohlen. Im Zusammenhang mit Thrombophilie ist die Untersuchung zum gegenwärtigen Zeitpunkt nicht Bestandteil der vertragsärztlichen Versorgung.
Erkrankung | ICD—10 | Gen | OMIM—G |
| Methylentetrahydrofolatreduktase- (MTHFR-) Defizienz | E72.1 | MTHFR | 607093 |
Hickey et al. 2013, Genet Med 15:153 / Dean, L., 2012 [Updated 2016] In: Pratt V, McLeod H, Dean L, et al., editors. Medical Genetics Summaries [Internet] / Cullen et al. 2009, J Lab Med 33:283 / Ulvik et al. 2007, Hum Genet, 121:57
letzte Aktualisierung: 9.4.2024