Wilson Erkrankung
Die Wilson-Krankheit ist eine seltene, autosomal-rezessive Stoffwechselerkrankung, die durch pathogene Varianten im ATP7B-Gen verursacht wird. Dieses Gen kodiert eine ATPase, die in Leber und Niere für die Bindung von Kupfer verantwortlich ist. Bei Patienten mit Wilson-Krankheit ist diese Kupferbindungsregion gestört. Trotz einer erhöhten Ausscheidung von Kupfer über die Nieren steigt die Menge an freiem Kupfer im Serum an, was zytotoxisch wirkt. Die Krankheit manifestiert sich bereits in den ersten Lebenstagen oder -wochen und führt in zwei Dritteln der Fälle im ersten Lebensjahr zum Tod. Patienten, die nicht von der akuten Phase betroffen sind, zeigen im Durchschnitt im Alter von 20 Jahren Symptome.
Wissenschaftlicher Hintergrund
Bei der Wilson Erkrankung handelt es sich um eine seltene autosomal-rezessive Erkrankung des Kupferstoffwechsels mit einer Prävalenz von 1:7.000-1:30.000. Erstmanifestationen der Erkrankung sind bereits in den ersten Lebenstagen oder -wochen zu beobachten (Dyspnoe, Apnoe sowie Zyanose). In 2/3 der Fälle führt die Erkankung bereits im ersten Lebensjahr zum Tod. Nicht von der lebensbedrohlichen, akuten Phase betroffene Patienten präsentieren sich im Durchschnitt im Alter von 20 Jahren mit Symptomen.
Ursache der Erkrankung sind pathogene Varianten im ATP7B-Gen, wodurch die Kupferbindungsregion einer ATPase in Leber und Niere gestört ist. Es kommt bei vermindertem Coeruloplasmin trotz gesteigerter renaler Cu2+-Ausscheidung zu einer Erhöhung des freien Serumkupfers, das im Gewebe zytotoxisch wirkt.
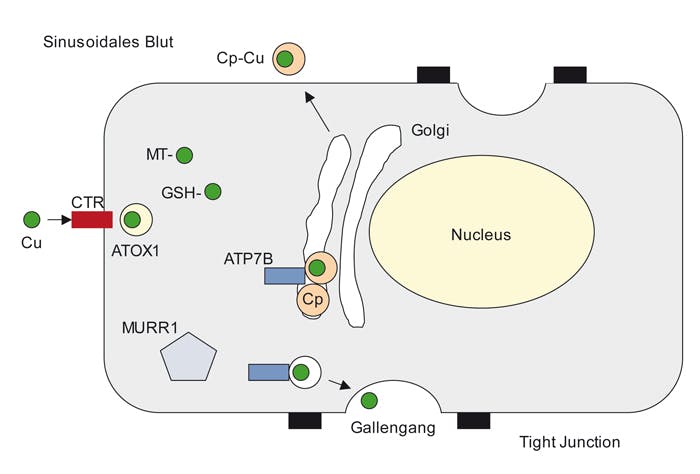
Ein typisches klinisches Merkmal dieser Erkrankung ist der goldbraun-grüne Kayser-Fleischer-Kornealring. Häufig sind auch neurologisch-psychiatrische Symptome in der Adoleszenz, wie z.B. Parkinson-ähnlicher Rigor und Tremor. Da wirksame Therapeutika existieren, welche die Prognose deutlich verbessern, ist die Frühdiagnose des Leidens von besonderer Bedeutung. Bei jeder unklaren Lebererkrankung, die vor dem 35. Lebensjahr auftritt, sollte eine Wilson-Erkrankung ausgeschlossen werden. Laborchemisch ist das Coeruloplasmin im Serum erniedrigt, die Kupfer-Ausscheidung im Urin erhöht. Ca. 1/3 der Patienten mit Wilson-Erkrankung zeigen eine Transversion von Cytosin nach Adenin, die zu einem His1069Gln-Aminosäureaustausch führt. Diese Variante (Allelfrequenz ca. 0,3% in der kaukasischen Bevölkerung) ist mit einem relativ milden Phänotyp und verzögerter Manifestation der Symptome assoziiert.
Erkrankung | ICD—10 | Gen | OMIM—G |
| Wilson Erkrankung (WND) | E83.0 | ATP7B | 606882 |
Hermann & Huster 2018, Nervenarzt 89:115; Weiss KH. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® (Updated 2016); Coffey et al. 2013, Brain 136:1476; Moller et al. 2011, Eur J Hum Genet 19:335; Mak et al. 2008, J Hum Genet 53:55
letzte Aktualisierung: 5.11.2023