Morbus Pompe
Der autosomal-rezessiv vererbte Morbus Pompe ist eine lysosomale Glycogen-Speicherkrankheiten, bei der es durch eine herabgesetzte Aktivität des Enzyms 1,4-α-Glukosidase zu einer Störung des Glycogenabbaus in den Lysosomen kommt, die vorwiegen Herz- und Muskelzellen vom Typ II betrifft. Ursächlich sind pathogene Varianten im GAA-Gen.
Wissenschaftlicher Hintergrund
Der autosomal-rezessiv vererbte Morbus Pompe (oder auch Glykogenose Typ II) zählt zu den lysosomalen Glycogen-Speicherkrankheiten. Durch eine herabgesetzte Aktivität des Enzyms 1,4-α-Glukosidase (GAA, saure Maltase) kommt es zur Störung des Glycogenabbaus in den Lysosomen. Ursächlich hierfür sind pathogene Varianten im GAA-Gen.
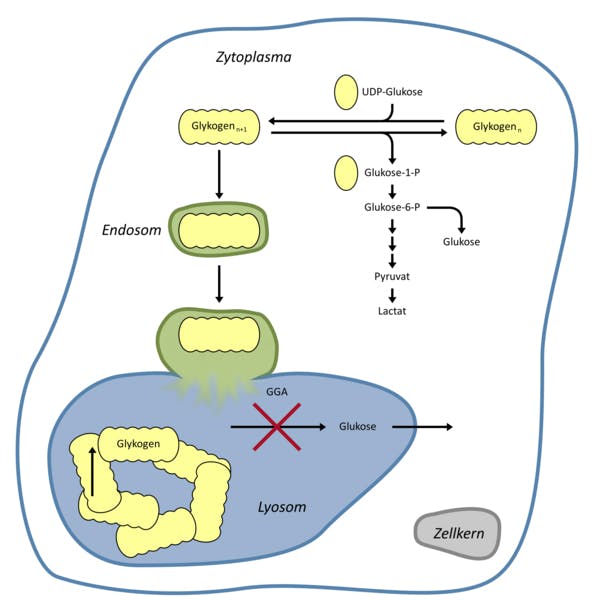
Der Enzymdefekt führt zu einer fortschreitenden systemischen Akkumulation von Glykogen in den Lysosomen. Vorwiegend sind Herzzellen und Muskelzellen vom Typ II betroffen. Der Morbus Pompe wird anhand des Auftretens der ersten Krankheitssymptome in zwei Formen eingeteilt: bei der infantilen Verlaufsform liegt ein homogenes klinisches Bild mit ausgeprägter Muskelschwäche (“floppy infants”), Ateminsuffizienz und Entwicklungsverzögerung vor. Unbehandelt sterben die meisten Kinder innerhalb des ersten Lebensjahres an Herzversagen.
Bei der late-onset Form, deren Verlauf hoch variabel ist, ist eine Abgrenzung zu anderen Muskelerkrankungen schwierig. Zu den Symptomen können fortschreitende Muskelschwäche, v.a. der rumpfnahen Muskulatur, Rückenschmerzen, Skoliose, Dyspnoe, Schlafapnoe, morgendliche Kopfschmerzen und Tagesmüdigkeit zählen.
Vor einer molekulargenetischen Untersuchung des GAA-Gens sollte eine Enzymaktivitätsbestimmung der 1,4-α-Glukosidase erfolgen. Die Erkrankung wird autosomal-rezessiv vererbt, d.h. beide Allele müssen betroffen sein, damit sich der Enzymmangel klinisch ausprägt. Es handelt sich um eine seltene Erkrankung mit einer weltweiten Inzidenz von ca. 1:40.000. Neben der symptomatischen Behandlung besteht seit 2006 in Europa die Möglichkeit einer Enzymersatztherapie.
Erkrankung | ICD—10 | Gen | OMIM—G |
| Morbus Pompe | E74.0 | GAA | 606800 |
Tarnopolsky et al. 2016, Can J Neurol Sci 43:472-85 / ACMG Practice Guideline, Wang et al. 2011, Genetics In Medicine 13:457 / Parkinson-Lawrence et al. 2010, Physiology, 25:102 / Schoser 2007, Akt Neurol 34:283 / ACMG Practice Guideline, Kishnani et al. 2006,Genetics IN Medicine 8:267 / Vellodi 2004, Brit J of Haematology, 128:413
letzte Aktualisierung: 5.11.2023