Fruktose-Intoleranz hereditäre (FBP1-Defizienz inbegriffen)
Die hereditäre Fruktose-Intoleranz (HFI) ist eine Stoffwechselerkrankung, die sich zumeist ab dem Säuglingsalter manifestiert und bei Aufnahme von Fruchtzucker zu schweren Hypoglykämien mit Zittern, Schweißausbrüchen und komatösen Zuständen führen kann. Sie wird durch ursächliche Varianten im ALDOB-Gen bedingt. Ähnliche Symptome treten bei einem Fruktose-1,6-bisphosphatase-(FBP1-) Mangel. auf, der durch Varianten im FBP1-Gen verursacht wird. Die HFI ist differentialdiagnostisch von der Fruktose-Malabsorption zu unterscheiden, die nicht mit der Untersuchung des ALDOB- oder FBP1-Gens abgeklärt werden kann.
Wissenschaftlicher Hintergrund
Bei der hereditären Fruktose-Intoleranz (HFI) handelt es sich um eine schwere autosomal-rezessiv vererbte Stoffwechselerkrankung, die sich in der Regel bereits im Säuglingsalter bei Zufütterung von Fruchtzucker manifestiert. Die HFI ist differentialdiagnostisch von der Fruktose-Malabsorption zu unterscheiden, die vorwiegend mit gastro-intestinalen Symptomen einhergeht.
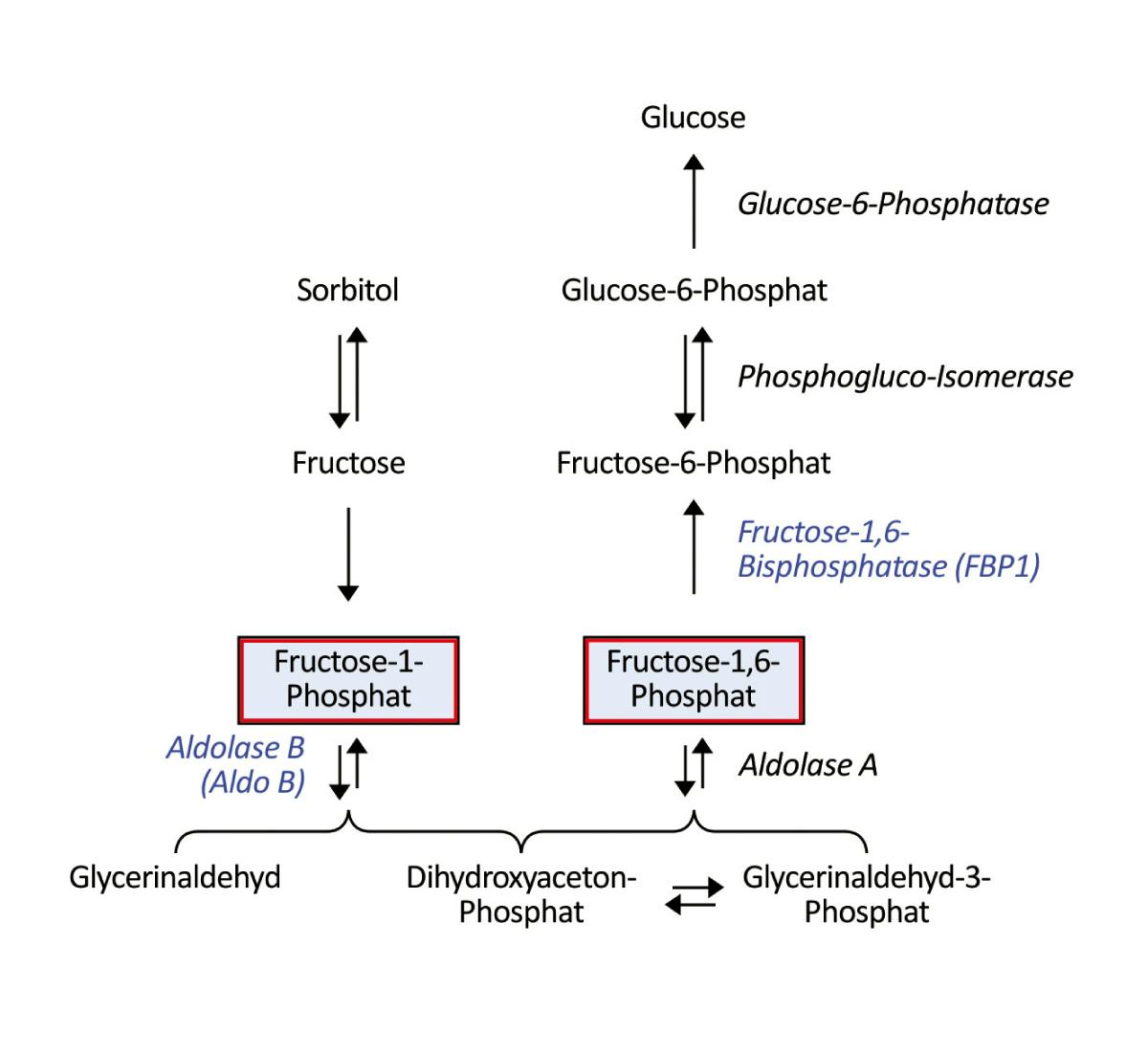
Die hereditäre Fruktose-Intoleranz führt bei Aufnahme von Fruchtzucker zu schwerwiegenden klinischen Symptomen. Bei chronischer Exposition kommt es zu Gedeihstörung und Leberzirrhose. Betroffene entwickeln häufig eine Abneigung gegen Süßes, wodurch die Erkrankung latent bleiben kann. Pathogene Varianten im Fruktaldolase B- (ALDOB-) Gen führen zu einem Enzymmangel, durch den es bei Fruktose-Zufuhr zur Akkumulation des toxischen Metaboliten Fruktose-1-Phosphat kommt. Durch die dadurch ausgelöste Hemmung der Gluconeogenese und Glycogenolyse treten Hypoglykämien mit Zittern, Schweißausbrüchen und komatösen Zuständen sowie Magen-Darm-Beschwerden auf. Vier häufige Varianten im ALDOB-Gen (p.(Ala150Pro), p.(Ala175Asp), p.(Asn335Lys) und die Stoppmutation p.(Tyr204*)) sind für ca. 85% der HFI-Fälle in der europäischen Bevölkerung verantwortlich, die Varianten p.(Ala150Pro) und p.(Ala175Asp) sind dabei in über 50% der Fälle ursächlich. Die Erkrankung wird autosomal-rezessiv vererbt, d.h. beide Allele müssen betroffen sein, damit sich der Enzymmangel klinisch ausprägt. Die Erkrankung ist mit einer Inzidenz von ca. 1:20.000 in der westlichen Bevölkerung eine seltene Erkankung.
Eine weitere Form der Fruchtzuckerunverträglichkeit mit HFI-ähnlichen Symptomen ist der hereditäre Fruktose-1,6-bisphosphatase-(FBP1-) Mangel.
Bleiben HFI oder FBP1-Mangel unerkannt und unbehandelt, können v.a. im Säuglingsalter potentiell lebensbedrohliche Situationen und im weiteren Verlauf progrediente Organschäden auftreten. Darüber hinaus kann die Behandlung mit Fruktose-, Saccharose- oder Sorbitol-haltigen Medikamenten zu schweren Nebenwirkungen führen.
Fruktose-Unverträglichkeiten können auch Folge von Störungen des Fruktose-Transports im Darm (Fruktose-Malabsorption) sowie chronisch entzündlichen Darmerkrankungen (z.B. M. Crohn) sein (sekundäre Form). Diese deutlich häufigeren Formen können in jedem Lebensalter auftreten und beruhen nicht auf einer genetisch bedingten Störung des Fruktose-Stoffwechsels. Die Magen-Darm-Symptomatik steht hier eindeutig im Vordergrund. Um gezielte Diagnostik zu gewährleisten, ist es wichtig, im Vorfeld einer genetischen Untersuchung beide Formen voneinander abzugrenzen.
Tran 2017, Nutrients. 9 pii:E356 / Bijarnia-Mahay et al. 2015, JIMD Rep 19:85 / Ferri et al. 2012, JIMD Rep 6:31 / Grochota et al. 2006, Mol Gen Metab 87:376 / Santer et al. 2005, Hum Mut 6:594 / Sanchez-Gutierrez et al. 2002, J Med Gen 39:5
letzte Aktualisierung: 4.11.2023