Adrenogenitales Syndrom (AGS)
Beim Adrenogenitalen Syndrom (AGS) handelt es sich um einen autosomal-rezessiv vererbten Mangel an Cortisol und gegebenenfalls Aldosteron. Bei der Erkrankung wird klinisch zwischen dem kongenitalen bzw. klassischen AGS und dem late-onset bzw. nicht-klassischen AGS unterschieden. Zumeist sind pathogene Varianten im 21-Hydroxylase-Gen (CYP21A2) ursächlich, (AGS Typ 3) jedoch können auch Veränderungen in anderen Genen zu einem AGS führen.
Wissenschaftlicher Hintergrund
Beim Adrenogenitalen Syndrom (AGS) handelt es sich um einen autosomal-rezessiv vererbten Mangel an Cortisol und ggf. Aldosteron mit einer Prävalenz von etwa 1:10.000 – 1:16.000 (kongenitales AGS) bzw. 1:500 – 1:1.000 (late-onset AGS). Die Erkrankung wird überwiegend durch pathogene Varianten im 21-Hydroxylase-Gen (CYP21A2) auf Chromosom 6p21.3 verursacht. Klinisch unterscheidet man das kongenitale bzw. klassische AGS vom late-onset bzw. nicht-klassischen AGS.
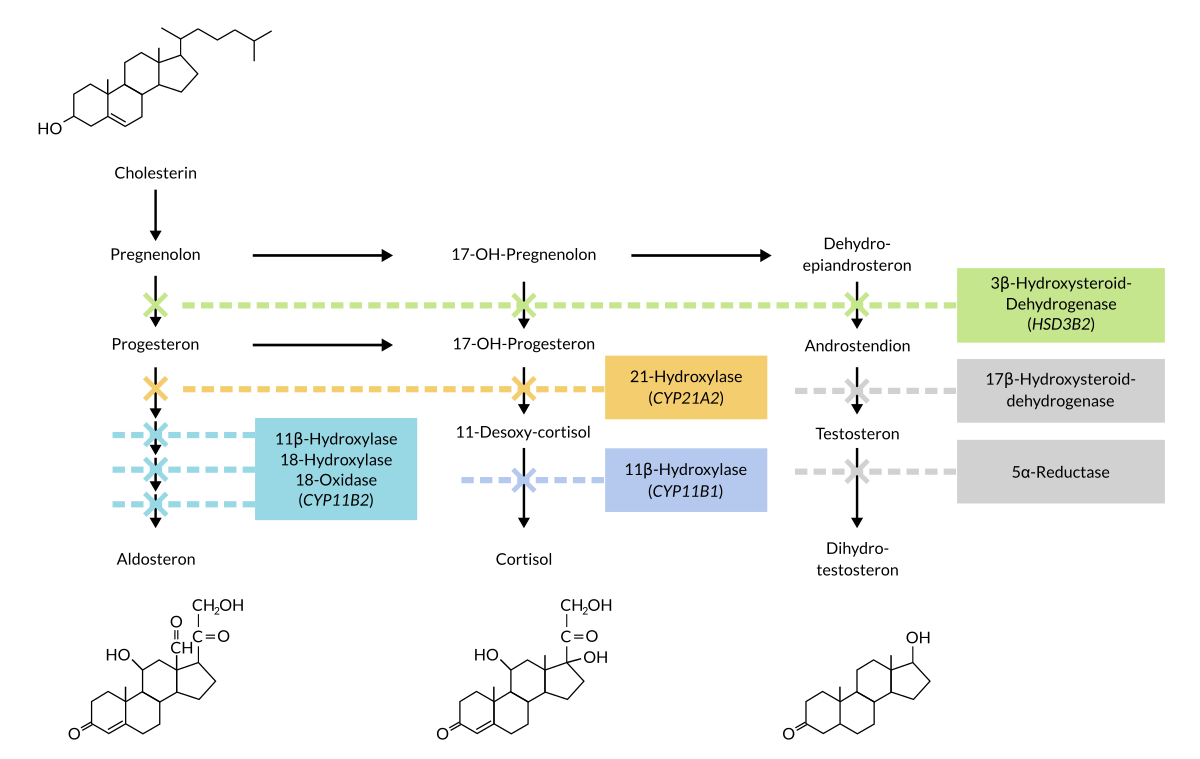
Das klassische AGS wird durch Varianten hervorgerufen, die zu einer hochgradigen Verminderung der 21-Hydroxylase-Enzymaktivität führen. Hierdurch kommt es zu einer stark verminderten enzymatischen Hydroxylierung von 17-OH-Progesteron und damit zu einem Mangel an Cortisol. Bei Vorliegen einer sehr schweren Enzymdefizienz kommt es durch die gestörte Hydroxylierung von Progesteron zusätzlich zu einem Aldosteronmangel. Der Stoffwechselblock führt über ein negatives Feedback zu einer vermehrten Ausschüttung von ACTH, wodurch es sekundär zu einer Nebennierenrindenhyperplasie mit Bildung männlicher Steroidmetaboliten und zur Störung der weiblichen Geschlechtsdifferenzierung kommt. Bei den betroffenen Mädchen kommt es bereits pränatal zu einer Virilisierung und der Ausbildung eines intersexuellen Genitales. Zusätzlicher Aldosteronmangel führt unbehandelt zu einem lebensbedrohlichen Salzverlustsyndrom. Betroffene Jungen können ebenfalls ein Salzverlustsyndrom zeigen; später kann eine Pubertas praecox vorliegen. Betroffene Kinder fallen im Verlauf durch eine beschleunigte Skelettreifung auf, wodurch sie zunächst zu groß, dann jedoch aufgrund des vorzeitigen Schlusses der Epiphysenfugen zu klein sind. Bei bekannter Anlageträgerschaft der Eltern kann der Virilisierung weiblicher Feten durch Dexamethasongabe während der Schwangerschaft vorgebeugt werden.
Beim nicht-klassischen oder late-onset AGS liegt eine weniger ausgeprägte Hyperandrogenämie vor, die sich vor allem bei erwachsenen Frauen u.a. mit Hirsutismus, Zyklusstörungen, tiefer Stimmlage und Akne manifestieren kann. Verursacht wird das late-onset AGS durch Homozygotie einer „milden pathogenen Variante“ oder durch kombinierte Heterozygotie einer „milden“ und einer „schweren pathogenen Variante“ bzw. zweier „milder pathogener Varianten“ im 21-Hydroxylase-Gen.
Pathogene Varianten im 11-β-Hydroxylase-Gen (CYP11B1) sind in ca. 5-8% aller klassischen Fälle des AGS ursächlich. Der daraus resultierende Stoffwechselblock führt ebenfalls zu einer vermehrten Bildung männlicher Steroidmetaboliten und zur Störung der weiblichen Geschlechtsdifferenzierung. Ein Salzverlustsyndrom tritt in der Regel nicht auf. In einzelnen Fällen konnten auch bei Frauen mit einem late-onset AGS Varianten im CYP11B1-Gen nachgewiesen werden.
Zudem kann die Analyse der Gene CYP11B2 (Aldosterone-Synthase), HSD3B2 (3β-Hydroxysteroid-Dehydrogenase)und CYP17A1 (Steroid-17α-Hydroxylase) durchgeführt werden.
Erkrankung | OMIM-P | ICD-10 | Gen | OMIM-G | Vererbung | Häufigkeit | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AGS Typ 3, Adrenogenitales Syndrom durch 21-Hydroxylase-Mangel | 201910 | E25.09 | CYP21A2 | 613815 | AR | ca. 95% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AGS Typ 4, Adrenogenitales Syndrom durch 11ß-Hydroxylase-Mangel | 202010 | E25.09 | CYP11B1 | 610613 | AR | ca. 5% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AGS Typ 2, Adrenogenitales Syndrom durch 3ß-Hydroxysteroiddehydrogenase-Mangel | 201810 | E25.09 | HSD3B2 | 613890 | AR | <1% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Nimkarn S, Gangishetti PK, Yau M, et al. 2002 [Updated 2016 Feb 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019 / New et al. 2013, Proc Natl Acad Sci USA 110:2611 / Speiser & White 2003, N Engl J Med 349:776
letzte Aktualisierung: 23.4.2024