Epilepsien
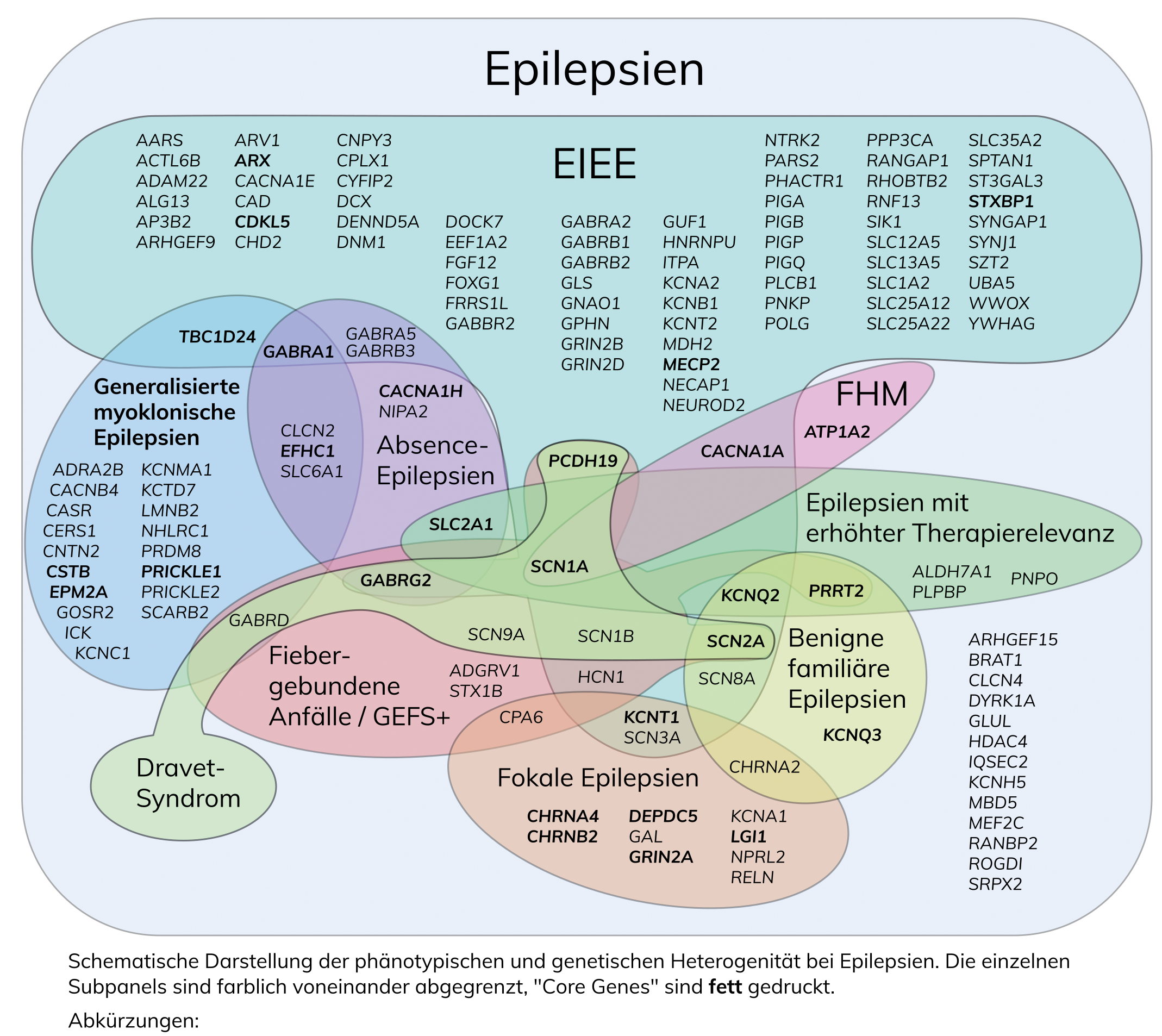
Epilepsien, mit einer Prävalenz von 0,5 bis 1%, beginnen oft im Kindesalter und sind zu mindestens 50% genetisch bedingt, meist aufgrund multifaktorieller oder polygener Ursachen. Nur ein kleiner Anteil der idiopathischen Epilepsien folgt einem monogenen Erbgang. Genetische Epilepsien sind häufig durch pathogene Varianten in Ionenkanälen oder Rezeptoren wie GABAA- und Nikotin-Acetylcholin-Rezeptoren verursacht, während symptomatische Epilepsien oft Folgen genetischer Syndrome oder Gehirnfehlbildungen sind. Genetische Diagnostik, insbesondere mittels Next Generation Sequencing (NGS), spielt eine wichtige Rolle in der Differenzialdiagnose und Therapieplanung von Epilepsien.
Wissenschaftlicher Hintergrund
Epilepsien treten mit einer Häufigkeit von 0,5 bis 1% auf, und knapp die Hälfte beginnt bereits im Kindesalter. In der heterogenen Gruppe der Epilepsien sind mindestens 50% genetisch bedingt, wobei die Ursache in den meisten Fällen multifaktoriell bzw. polygen ist. Nur 1 bis 2% der sog. idiopathischen Epilepsien folgt einem monogenen Erbgang. Zu diesen gehören unter anderem die epileptischen Enzephalopathien des Kindesalters (EIEE), die früh beginnen, einen schweren Verlauf zeigen, oft therapieschwierig sind und neben der fast immer vorhandenen Störung der kognitiven Entwicklung weitere Komorbiditäten zeigen. Bei den idiopathischen generalisierten Epilepsien beispielsweise erlaubt der Nachweis einer pathogenen Variante in einem der aufgeführten Gene allerdings häufig nur den Schluss auf ein erhöhtes Risiko, eine Epilepsie zu entwickeln (Suszeptibilitätsfaktoren). Ein Großteil der genetischen Epilepsien ist durch pathogene Varianten in Untereinheiten neuronaler spannungsabhängiger Na+ , K+ , Ca2+ , Cl– -Ionenkanäle und Untereinheiten ligandenabhängiger Rezeptoren wie dem GABAA -Rezeptor und dem Nikotin-Acetylcholin-Rezeptor bedingt.
Den genetischen Epilepsien können die symptomatischen Epilepsien gegenüber gestellt werden, die z.B. sekundär als Folge einer angeborenen Gehirnfehlbildung (z.B. Migrationsstörung), im Rahmen eines übergeordneten genetischen Syndroms (z.B. Angelman-Syndrom, Rett-Syndrom, Tuberöse Sklerose) oder bei chromosomalen Imbalancen auftreten.
Die klinische Differenzialdiagnose kann oft schwierig sein, weshalb die genetische Diagnostik auch mittels NGS zunehmend eine Möglichkeit der Ursachenklärung darstellt. Der Nachweis einer pathogenen Variante kann eine Verdachtsdiagnose bestätigen, was bei einigen Formen eine gezielte Therapie ermöglicht (z.B. beim Glucose-Transporter-Defekt). Zudem kann weitere Diagnostik eingespart, eine prognostische Einschätzung und Aussagen zu einem eventuellen Wiederholungsrisiko gegeben werden.
Myers et al. 2019, Clin Genet 95(1): 95 / Scheffer et al. 2017, Epilepsia; 58(4):512 / Lesca et al. 2015, Rev Neurol (Paris) 6-7:539 / Poduri et al. 2014, Nat Rev Neurol 10(5):293 / Scheffer 2014, Neuropediatrics 45:70 / Thomas et al. 2014, Nat Rev Neurol 10:283 / Thomas et al. 2014, Nat Rev Neurol 10:283 / Hoppman-Chaney et al. 2012, Clin Genet 83:345 / Tavyev Asher et al. 2012, Eur J Med Genet 55:299 / Striano et al. 2012, Arch Neurol 69:322 / Noh et al. 2012, Eur J Med Genet 55:281 / Weber et al. 2011, Z Epileptol 24:100 / Nicita et al. 2011, Seizure 21:3 / Muhle et al. 2011, Epilepsia 52:e194 / Depienne et al. 2010, Hum Mut 32:E1959 / Berg et al. 2010, Akt Neurol 37:120 / OMIM, Online Mendelian Inheritance In Man
letzte Aktualisierung: 24.3.2024