Muskeldystrophie Duchenne / Becker
Muskeldystrophien sind genetische Muskelerkrankungen, wobei die Muskeldystrophie Duchenne (DMD) die häufigste bei männlichen Neugeborenen ist, sich zwischen dem 3. und 5. Lebensjahr manifestiert und zu einem fortschreitenden Muskelabbau führt. Die Betroffenen werden oft bis zum 12. Lebensjahr rollstuhlpflichtig und haben eine reduzierte Lebenserwartung von 20-30 Jahren. Die Muskeldystrophie Becker (BMD) ist milder und erlaubt meist das Gehen bis zum 60. Lebensjahr. Beide Erkrankungen resultieren aus pathogenen Varianten im Dystrophin-Gen, die bei DMD zu einem verkürzten, inaktiven Polypeptid führen, während der BMD eine reduzierte Synthese oder ein strukturell verändertes Protein mit Restaktivität zugrunde liegt.
Wissenschaftlicher Hintergrund
Bei den Muskeldystrophien handelt es sich um eine klinisch und genetisch heterogene Gruppe von Muskelerkrankungen. Die Einteilung erfolgt nach dem Vererbungsmodus in autosomal-dominante (Fazioskapulohumerale), autosomal-rezessive (Gliedergürtel) und X-chromosomal-rezessive (Duchenne/Becker) Muskeldystrophien.
Die Muskeldystrophie Duchenne (DMD) ist mit einer Prävalenz von 1:3.500 (männliche Neugeborene) die häufigste Muskelerkrankung im Kindesalter. Sie manifestiert sich im 3. – 5. Lebensjahr mit einer Schwäche der Becken- und Oberschenkelmuskulatur. Typisch sind zudem ein watschelnder Gang, eine Wadenhypertrophie sowie ein positives Gowers-Zeichen (Hochklettern an sich selbst). Der Erkrankungsverlauf ist progredient, so dass die meisten Betroffenen im Alter von ca. 12 Jahren rollstuhlpflichtig werden. Zusätzlich sind auch die Atem- und die Herzmuskulatur betroffen. Die Lebenserwartung ist deutlich verkürzt und liegt bei etwa 20-30 Jahren. Die milder verlaufende Muskeldystrophie Becker (BMD) tritt mit einer Häufigkeit von 1:20.000 (männliche Neugeborene) auf und betrifft ebenfalls die Becken- und Oberschenkelmuskulatur. Die Erkrankung verläuft aber nach der Manifestation zwischen dem 6. und 12. Lebensjahr wesentlich günstiger, so dass die Gehfähigkeit zumeist bis zum 60. Lebensjahr erhalten bleiben kann. Klinisch kommt es sowohl bei der DMD als auch bei der BMD zu einem fortschreitenden Abbau der Muskelzellen, der mit einer erhöhten Serum-Creatin-Kinase (CK) auf das bis zu 100-fache des Normalwertes einhergeht. Auch bei Überträgerinnen einer DMD oder BMD kann der CK-Wert erhöht sein. Neben der Serum-CK kann auch ein Elektromyogramm oder die histologische Untersuchung einer Muskelbiopsie zur Diagnosestellung herangezogen werden.
Pathogene Varianten im Dystrophin (DMD)–Gen sind die genetische Ursache der X-chromosomal vererbten Muskeldystrophien Duchenne (DMD) und Becker (BMD). Beim Dystrophin handelt es sich um ein Protein des Zytoskeletts, das an der Sarkolemm-Membran der Skelettmuskelfasern lokalisiert ist. Bei DMD-Patienten kommt es infolge pathogener Veränderungen zur Synthese eines verkürzten, inaktiven Polypeptids, welches vorzeitig abgebaut wird. Ursache dafür sind Deletionen oder Duplikationen, die zu einer Verschiebung des Leserasters führen (out-of-frame) sowie translationale Stopvarianten infolge von Nonsense-Varianten, kleinen Deletionen, Insertionen und Spleißvarianten. Bei BMD-Patienten ist die Dystrophin-Bioynthese entweder stark reduziert oder es wird ein verkürztes oder strukturell verändertes Protein mit Restaktivität produziert. In frame-Deletionen und -Duplikationen, die nicht zu einer Verschiebung des Leserasters führen, sowie Punktmutationen außerhalb der funktionellen N- und C-terminalen Domäne führen zum milderen BMD-Phänotyp. Etwa 30% der Betroffenen erkranken aufgrund einer pathogenen de novo Variante. In ca. 70% handelt es sich um familiäre Fälle, bei denen die Mutter Überträgerin der Erkrankung ist. 65% aller pathogenen Varianten bei DMD und 85% derer bei BMD sind Deletionen einzelner oder mehrerer Exons. 6-10% aller pathogenen Varianten bei BMD und DMD sind Duplikationen. Für beide Variantentypen gibt es proximale und distale Hotspot-Regionen. 25-30% aller DMD-Patienten und 5-10% aller BMD-Patienten weisen Punktmutationen, Spleißvarianten oder kleinere Insertionen bzw. Deletionen innerhalb des DMD-Gens auf, die über das gesamte Gen verteilt sein können.
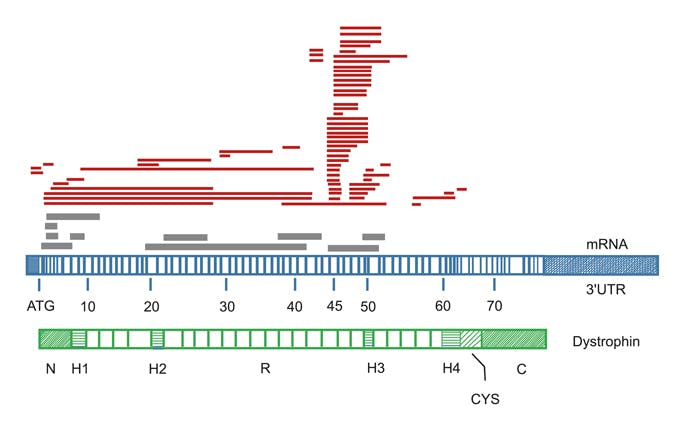
Abb.: Schematische Darstellung der Dystrophin-mRNA und des Dystrophin-Proteins. Rot: Lokalisation der identifizierten Deletionen, grau: Duplikationen. Hotspots für Deletionen sind Exon 45-52, für Duplikationen der N-Terminus (modifiziert nach Worton et al.).
Erkrankung | ICD—10 | Gen | OMIM—G |
| Muskeldystrophie Duchenne / Becker | G71.0 | DMD | 300377 |
Allen et al. 2018 Ped 141:e20172391 / Okubo et al. 2017, Orpht J of Rare Dis 12:149 / Tuffery-Giraud et al. 2009, Hum Mutat 30:934 / Ashton et al. 2008, Eur J Hum Genet 16:53 / Aartsma-Rus et al. 2006, Muscle Nerve 34:135 / Lalic et al. 2005, Eur J Hum Genet 13:1231 / Flanigan et al. 2003, Am J Hum Genet 72:931 / van Essen et al. 1997, Med Genet 34:805 / Gillard et al. 1989, Am J Hum Genet 45:507 / Koenig et al. 1989, Am J Hum Genet 45:498 / http://www.dmd.nl
letzte Aktualisierung: 26.10.2023