Core-Myopathien (bzw. RYR1-assoziierte Myopathien) und Nemaline Myopathien
Kongenitale Myopathien sind eine seltene, heterogene Gruppe von Muskelerkrankungen, die typischerweise von Geburt an vorliegen und sich durch Muskelschwäche und -schwund auszeichnen. Diese Krankheiten können durch pathogene Varianten in über 20 verschiedenen Genen verursacht werden, wobei das RYR1-Gen die häufigste genetische Ursache darstellt. Zu den bekanntesten kongenitalen Myopathien zählen die Central core disease (CCD), centronukleäre Myopathie und Nemaline Myopathie. Insbesondere Patienten mit CCD können eine Neigung zur Malignen Hyperthermie zeigen, einer potenziell lebensbedrohlichen Reaktion auf bestimmte Narkosemittel.
Wissenschaftlicher Hintergrund
Die kongenitalen Myopathien beschreiben eine eigene Krankheitsentität unter den Myopathien, die in der Regel von Geburt an vorliegen und meist langsam oder nicht progredient verlaufen. Auch schwere, fatale Formen und spätmanifestierende Formen des Erwachsenenalters sind bekannt. Es handelt sich um eine seltene, heterogene Krankheitsgruppe mit einer geschätzten Prävalenz von 1:25.000 und einem äußerst variablen Symptomenspektrum. Bisher sind pathogene Varianten in mehr als 20 Genen bekannt, die zur Ausprägung einer der Muskelerkrankungen aus dieser Gruppe führen können.
Im Vordergrund stehen Muskelschwäche und –schwund, symmetrische, proximale Paresen ohne Sensibilitätsstörung. Historisch wurde versucht, die verschiedenen (Unter-)Formen anhand von biopsiertem Muskelgewebe zu klassifizieren. Strukturauffälligkeiten innerhalb der Muskelfasern, wie Strukturdefekte, abnorme Einschlüsse und/oder Kernanomalien können vorliegen. Allen gemeinsam ist ein myopathologisches Grundmuster der Fasertypendisproportion, d.h. numerische Prädominanz und selektive Hypotrophie der Typ-1-Fasern gegenüber der Norm (Reduktion des Durchmessers der Typ-I-Muskelfasern von mindestens 12 % im Vergleich zu den Typ-II-Muskelfasern). Eine eindeutige Klassifikation ist oft schwierig, einige Formen können durch pathogene Varianten in verschiedenen Genen verursacht werden oder aber Veränderungen in einem Gen können zu verschiedenen Typen führen. Oft wird die definitive Diagnose dennoch durch die molekulargenetische Untersuchung gestellt bzw. bestätigt.
Zu den häufigeren, länger bekannten und damit „klassischen“ kongenitalen Myopathien gehören die Central core disease (CCD), die centronukleäre Myopathie (CNM) und die Nemaline Myopathie. Andere histopathologische Varianten sind u.a.: Multiminicore disease (MmD), Kongenitale Fasertypendisproportion (CFTD) und das King Denborough-Syndrom.
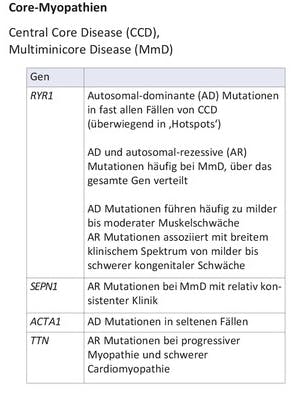
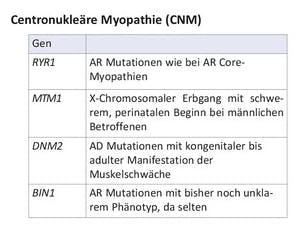
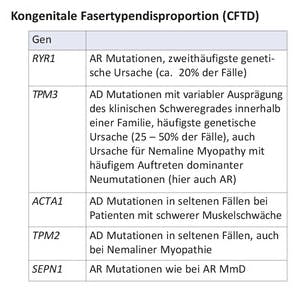
Autosomal-dominante und auch autosomal-rezessive pathogene Varianten im RYR1-Gen sind die häufigste Ursache der kongenitalen Myopathien. Das Genprodukt ist ein Calcium-freisetzender Kanal im Skelettmuskel, der mit Kopplung von Erregung und Kontraktion assoziiert ist. Dominante Varianten in diesem Gen stehen im Zusammenhang mit CCD und/oder Suszeptibilität für maligne Hyperthermie (MHS), während rezessive Varianten bei Patienten mit MmD, CNM und CFTD zu finden sind.
Die CCD präsentiert sich typischerweise ab der Geburt mit Hypotonie und motorischer Entwicklungsverzögerung. Dabei ist vor allem die proximale Muskulatur betroffen. In der Muskelbiopsie gelingt der Nachweis von meist zentral in Muskelfasern gelegenen, substratdefekten „Cores“ in der oxidativen Enzymfärbung aufgrund fehlender Mitochondrien. Von besonderer klinischer Bedeutung ist die Tatsache, dass Patienten mit Central-Core-Myopathie häufig eine Prädisposition zur Malignen Hyperthermie aufweisen, einer Erkrankung, die als lebensbedrohliche Komplikation einer Inhalationsnarkose in Erscheinung treten kann.
Witting et al. 2017 Neurol Genet 3:e140 / Neto et al. 2017, Neuromuscul Disord. 27:975 / North et al. 2014, Neuromuscular Disorders 24: 97 / Amburgey et al. 2013, Orph J of Rare Dis 8:117 / Klein et al. 2012, Hum Mut 33:61 / Amburgey et al. 2011, Ann Neurol 70:662 / Goebel et al. 2009, Pathologe 30:365 / Jungbluth 2007, Orph J of Rare Dis 2:25
letzte Aktualisierung: 18.3.2024