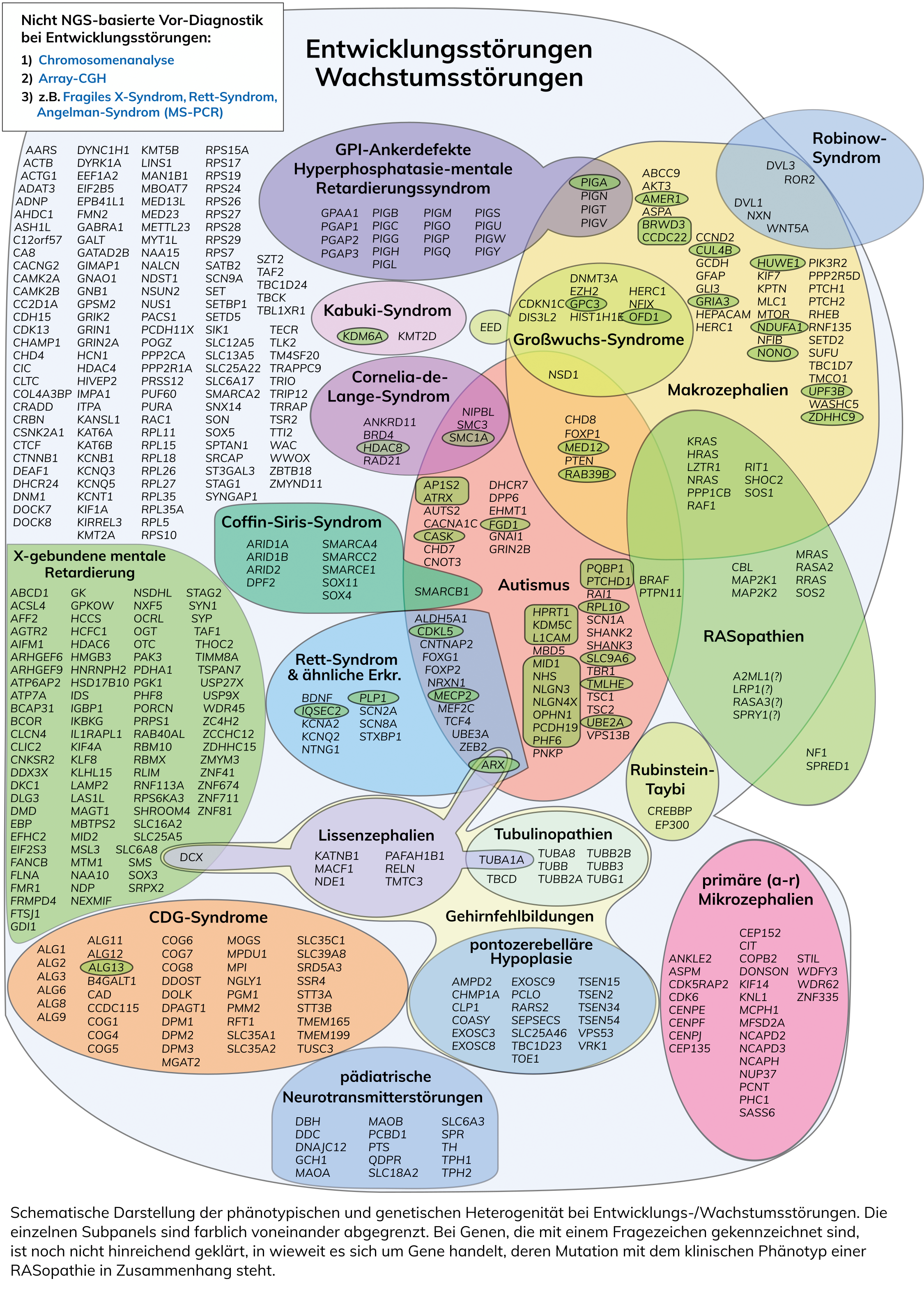
Entwicklungsstörungen
Intelligenzminderung, definiert durch einen IQ unter 70, betrifft 1,5 bis 2% der Bevölkerung, wobei schwerere Formen (IQ < 50) eine Prävalenz von 0,3 bis 0,4% haben. Genetische Faktoren tragen mindestens 50% zur Intelligenzminderung bei. Studien zeigen, dass bis zu 30% der schweren, nicht-syndromalen Entwicklungsstörungen durch de novo Punktmutationen und kleine Indels verursacht werden. Next Generation Sequencing (NGS) und Whole-Exome-Analyse (WES) sind fortschrittliche diagnostische Methoden, die eine breite Analyse genetischer Ursachen ermöglichen.
Wissenschaftlicher Hintergrund
Eine Intelligenzminderung, definiert als ein IQ von unter 70, hat eine Prävalenz von 1,5 bis 2%. Schwerere Formen mit einem IQ von < 50 haben eine Prävalenz von 0.3 bis 0,4%. Jungen/Männer sind aufgrund X- chromosomaler Gene häufiger betroffen. Die Ursachen einer Intelligenzminderung sind vielfältig; genetische Faktoren sind aber zu mindestens 50% beteiligt. Mit den bisher möglichen Untersuchungen wie Chromosomenanalyse, Array-CGH und einer Zieldiagnostik (z.B. Fragiles-X-Syndrom, Rett-Syndrom, Angelman-Syndrom) bleiben noch ca. 60% der Ursachen von Entwicklungsstörungen ungeklärt. Mehrere Studien der letzten Jahre, in denen Patienten mit schwerer Intelligenzminderung (IQ<50) mittels neuer Hochdurchsatz-Techniken (NGS) untersucht wurden, konnten bestätigen, dass dominante, pathogene de novo Varianten offenbar zu einem großen Teil zur Ursache der schweren Intelligenzminderung beitragen (z. B. Vissers L. et al, Nat Genet, 2010, de Ligt, J. et al, NEJM, 2012 und Rauch, A. et al, Lancet, 2012). Man geht nach diesen Studien davon aus, dass bis zu ca. 30% der schweren, nicht-syndromalen Entwicklungsstörungendurch de novo Punktmutationenund kleine Indels verursacht werden, wobei eine große genetische Heterogenität zu beobachten ist. Darüber hinaus spielen auch autosomal-rezessive Mutationen bei den Entwicklungsstörungen eine Rolle (ca. 13-24%), sowie Mutationen in X-chromosomalen Genen (5-10% der männlichen Betroffenen). Als weiterführende Diagnostik besteht die Möglichkeit der gleichzeitigen Analyse einer großen Anzahl von Genen, die mit neurologischen bzw. Entwicklungsstörungen in Zusammenhang stehen und die bereits in den Datenbanken gelistet sind, mittels Next Generation Sequencing (NGS).
Bei einer Reihe von Entwicklungsstörungen stehen auch andere Symptome im Vordergrund, so z.B. Autismus-Spektrum-Störungen oder auffällige Wachstumsparameter, z.B. Makrozephalie, Mikrozephalie oder Großwuchs. Darüber hinaus gibt es syndromale Formen von Entwicklungsstörungen, bei denen differenzialdiagnostisch weitere Syndrome und damit weitere ursächliche Gene in Frage kommen (z.B. Rett- und Rett-ähnliche-Syndrome). Schließlich sind hier auch einige der bekannteren, genetisch heterogenen Syndrome aufgeführt, die sinnvoll mittels NGS untersucht werden können (Cornelia-de-Lange-Syndrom, RASopathien u.a.).
Alternativ ist auch noch eine erweiterte Diagnostik mittels Whole-Exome-Analyse (WES) möglich.
Mir et Kuchay 2019, J of Med Genet doi: 10.1136/jmedgenet-2018-105821 / Jamra 2018, Med Genet 30:323 / Hu et al. 2019, Mol Psychiatry 24:1027 /Vissers et al 2016 Nat Rev Genet 17:9 / Grozeva et al Tzschach et al 2015 Eur J of Hum Genet 23:1513 / Tan et al 2015 Clin Genet. doi: 10.1111 / cge.12575 / Musante et al 2014 Trends Genet 30:32 / Brett et al 2014 PLoS One 9(4): e93409 / Redin et al 2014 J Med Genet. 2014 51:724
letzte Aktualisierung: 11.3.2024