Prader-Willi-Syndrom (PWS)
Das Prader-Willi-Syndrom (PWS) ist eine genetische Erkrankung mit einer Prävalenz von 1:15.000 bis 1:30.000, charakterisiert durch Muskelhypotonie, Gedeihstörungen im Säuglingsalter, Hyperphagie mit nachfolgender Adipositas und eine mäßige mentale Retardierung. Beim PWS ist eine Chromosomenregion (15q11.2-q13) betroffen, die dem Genomic Imprinting unterliegt, wobei etwa 70% der Fälle durch eine Mikrodeletion auf dem väterlichen Chromosom 15 und 30% durch eine maternale uniparentale Disomie 15 verursacht werden. Diagnostisch werden verschiedene Methoden eingesetzt, um Mikrodeletionen, Methylierungsveränderungen und uniparentale Disomien zu erfassen.
Wissenschaftlicher Hintergrund
Das Prader-Willi-Syndrom (PWS) zeichnet sich im Säuglingsalter durch eine ausgeprägte Muskelhypotonie mit Trinkschwäche und Gedeihstörung aus. Während der Schwangerschaft können verminderte Kindsbewegungen auffallen; die Geburt kann aus Beckenendlage erfolgen. Die motorische Entwicklung ist mäßig verzögert, die Kinder können mit durchschnittlich einem Jahr sitzen, mit zwei Jahren frei laufen. Es besteht eine mäßige mentale Retardierung, bei ca. 40% liegt die Intelligenz im unteren Normbereich, trotzdem sind Lernschwierigkeiten häufig. Jenseits des Säuglingsalters kommt es zur Hyperphagie mit Entwicklung einer Adipositas und nachfolgenden Komplikationen wie Diabetes mellitus und kardiopulmonalen Erkrankungen. Meist entwickelt sich ein Minderwuchs. Es besteht ein Hypogenitalismus bzw. Hypogonadismus mit niedrigen Hormonspiegeln; die Pubertätsentwicklung ist oft nicht altersgemäß. Als typische Verhaltensweisen werden Sturheit und Wutanfälle sowie Hautzupfen mit der Tendenz zur Selbstverletzung beobachtet. Gute Fähigkeiten zeigen Patienten mit Prader-Willi-Syndrom im Bereich der visuellen Erfassung und Verarbeitung wie z.B. beim Zusammenlegen von Puzzles. An äußeren Merkmalen findet man oft ein schmales Gesicht, mandelförmige Augen, Strabismus, einen kleinen Mund mit schmaler Oberlippe, kleine Hände und Füße. Bei den meisten Patienten mit einer Mikrodeletion als Ursache des Prader-Willi-Syndroms ist eine gewisse Hypopigmentierung festzustellen.
Die Häufigkeit wird mit 1:15.000 bis 1:30.000 angegeben. Die krankheitsverursachenden Gene liegen beim Prader-Willi-Syndrom (und dem Angelman-Syndrom) in einer Chromosomenregion (15q11.2-q13), die dem sog. Genomic Imprinting unterliegt. Diese elternspezifische Prägung bewirkt, dass sich Gene im Grad der DNA-Methylierung, der Chromatinstruktur und damit der Expression unterscheiden, je nachdem, von welchem Elternteil sie stammen. Die Steuerung erfolgt über ein zweigeteiltes Imprintingzentrum in 15q11.2-q13. Aufgrund dieser Besonderheit können das Prader-Willi-Syndrom und das Angelman-Syndrom neben der Mikrodeletion weitere Ursachen haben, die zum Expressionsverlust der betreffenden Gene führen. Mehrere Gene im Bereich 15q11.2-q13 werden nur vom väterlichen Chromosom 15 exprimiert und stehen in ursächlichem Zusammenhang mit dem Prader-Willi-Syndrom. Ca. 70% der PWS-Patienten haben eine Mikrodeletion 15q11.2-q13 auf dem vom Vater vererbten Chromosom 15. Rund 30% haben eine maternale uniparentale Disomie 15 (UPD), d.h. beide Chromosomen 15 stammen von der Mutter, keines vom Vater, ca. 1% haben eine Störung im Imprinting-Zentrum, bei wenigen wurde eine chromosomale Strukturaberration unter Einbeziehung der Region 15q11.2-q13 gefunden.
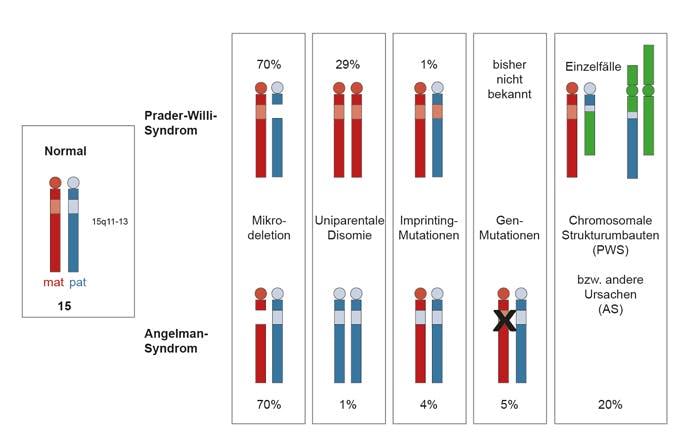
Es gibt gewisse Genotyp-Phänotyp-Korrelationen. Die zytogenetische (FISH-)Analyse erfasst nur die Mikrodeletionen, die methylierungssensitive MLPA erfasst Mikrodeletionen und Methylierungsveränderungen, kann jedoch die Grundlage: UPD bzw. Imprinting-Mutationen nicht spezifizieren. Die Mikrosatellitenanalyse erfasst eine maternale uniparentale Disomie 15 (UPD).
Butler et al. 2018, Am J Med Genet A 176(2):368 / Cassidy et al. 2009, Eur J Hum Genet 17:3 / Leitlinien der Deutschen Gesellschaft für Humangenetik und dem Berufsverband der Deutschen Humangenetiker e.V. 2010, medgen 22:282 / Sarimski 2003: Prader-Willi-Syndrom, in: Entwicklungspsychologie genetischer Syndrome, Hogrefe, 3.Aufl. / Rost 2000, Monatsschr Kinderheilkd 148: 55-69 / Zeschnigk et al. 1997, Eur J Hum Genet 5: 94-98 / Holm et al. 1993, Pediatrics 91:398-402
letzte Aktualisierung: 11.3.2024