Osteogenesis imperfecta
Die Osteogenesis imperfecta (OI) oder Glasknochenkrankheit ist eine Gruppe von Erkrankungen, die durch erhöhte Knochenbrüchigkeit gekennzeichnet sind und meist autosomal-dominant vererbt werden. Die häufigsten Formen sind auf Varianten in den COL1A1- und COL1A2-Genen zurückzuführen, die Typ I-Kollagen beeinflussen. Es gibt mehrere Typen von OI, die sich in Schweregrad und Symptomen unterscheiden, wobei genetische Varianten in verschiedenen Genen wie COL1A1, COL1A2, FKBP10, CRTAP, LEPRE1 und PPIB mit den verschiedenen Typen assoziiert sind. Die Erkrankung manifestiert sich in Symptomen wie Knochenbrüchen, Skelettdeformitäten und Hörverlust.
Wissenschaftlicher Hintergrund
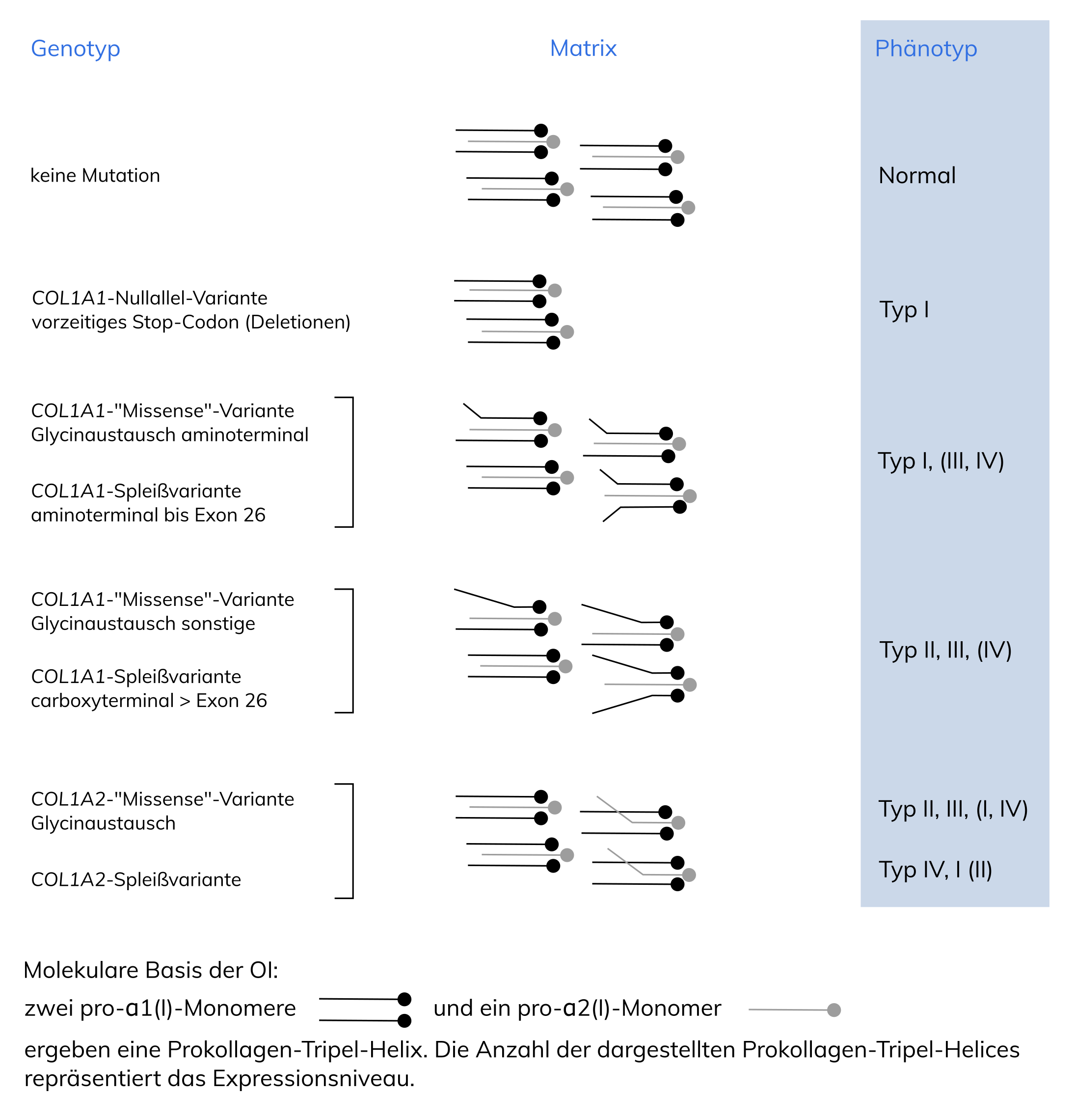
OI oder Glasknochenkrankheit (Häufigkeit etwa 1:10.000) ist eine klinisch und genetisch heterogene Gruppe von Erkrankungen mit erhöhter Knochenbrüchigkeit, die, abgesehen von sehr seltenen Sonderformen, autosomal-dominant vererbt werden. In 97% der OI-Typ I-Fälle und 77% der moderat bis schweren Formen können ursächliche Varianten in den COL1A1– und COL1A2-Genen nachgewiesen werden. Häufig führen diese zur Substitution von Glycin in der Tripel-Helix-Domäne des Typ I-Kollagens. In ca. 9% der moderat bis schweren Fälle sind Varianten im IFITM5-Gen ursächlich. Die klinische Symptomatik hängt vom betroffenen Gen sowie von Art und Lokalisation der Variante ab (Genotyp-Phänotyp-Korrelation).
Die seltenen autosomal-rezessiv vererbten Sonderformen der OI sind meist durch spezifische klinische Merkmale charakterisiert. Die Analyse dieser Gene wird derzeit in internationalen Leitlinien nur nach gründlicher klinischer Evaluierung empfohlen.
OI wird in folgende Erkrankungstypen klassifiziert:
1. Autosomal-dominant vererbte Formen (häufig)
Typ I (häufigste Form, ca. 65% d. Fälle) ist gekennzeichnet durch eine milde Verlaufsform mit mäßiger Knochenbrüchigkeit (10-20 Knochenbrüche bis zur Pubertät), Blauverfärbung der Skleren und postpubertärem Hörverlust bei 50% der Betroffenen. Auch Tinnitus, Aorteninsuffizienz und dünne Haut (in ca. 20% der Fälle) sind charakteristisch. Man unterscheidet Typ IA mit und Typ IB ohne Dentinogenesis imperfecta.
Typ II (ca. 20% d. Fälle) ist die schwerste Verlaufsform und verläuft meist intrauterin oder in den ersten Wochen postnatal letal. Sporadische Fälle werden oft durch Keimbahnmosaike verursacht, das Wiederholungsrisiko bei Folgeschwangerschaften beträgt ca. 10%.
Typ III (ca. 5% d. Fälle) ist phänotypisch besonders variabel. Typisch sind extreme Kleinwüchsigkeit, Skelettdeformitäten, ca. 100 Knochenbrüche bis zur Pubertät und Hörverlust. Weiche Knochen, Skoliose und Dentinogenesis Imperfecta sind ebenfalls charakteristisch.
Typ IV (ca. 10% der Fälle) ist eine milde Verlaufsform mit Kleinwüchsigkeit und mäßigen Skelettdeformitäten ohne Sklerenverfärbung, mit mäßiger Knochenbrüchigkeit. Man unterscheidet Subtyp A und B mit und ohne Dentinogenesis Imperfecta.
Die Erkrankung wird durch Varianten in den Typ I-Kollagen-Genen COL1A1 (2/3 d. Fälle) und COL1A2 (1/3 d. Fälle) verursacht, die zu einer verminderten Synthese von Prokollagen alpha1, alpha2 oder zu einer Strukturveränderung von Kollagen führen. Die phänotypische Heterogenität wird vermutlich durch den Einfluss modifizierender Gene und durch strukturelle Unterschiede verursacht. Insgesamt sind etwa 1.000 Varianten beschrieben, davon betreffen ca. 60% die Aminosäure Glycin.
Typ V ist mit wenigen Einzelfällen mit hypertropher Kallusbildung, maschenartiger Histologie der Knochen, dichten Epiphysen, Skelettdeformitäten und variabler Knochenbrüchigkeit unbekannter Ursache beschrieben.
2. Autosomal-rezessiv vererbte Formen (selten)
Typ VI ist an wenigen Einzelfällen mit mäßig schwerer Form der OI mit Skelettdeformitäten und variabler Knochenbrüchigkeit beschrieben. In der Histologie zeigen sich Fischgräten-artige Lamellen. Kürzlich konnten in wenigen Familien türkischer Abstammung Varianten im FKBP10-Gen nachgewiesen werden, das für das Chaperon FKBP65 codiert. FKBP65 ist an der Faltung von Typ I-Kollagen beteiligt.
Typ VII (2-3% der letalen OI-Fälle) ist charakterisiert durch multiple Knochenbrüche, extrem geringe Mineralisierung und “Popkorn-Epiphysen”. Die Ursache liegt in Varianten im CRTAP-Gen. CRTAP codiert einen Bestandteil des Kollagen 3-Hydroxylierungs-komplexes (post-translationale Prolyl-3-Hydroxylierung von Kollagen Typ I und II). Dieser modifiziert Pro986 der alpha1(I)-Kette, wodurch deren Faltung erleichtert und die Kette stabilisiert wird. Eine fehlende Pro986-Hydroxylierung führt zur verzögerten Faltung der Kollagen-Helix und deren Überschussmodifikation (28%-43% Zunahme der Hydroxylierung von Lysinresten). Da Typ II-Kollagen im Knorpel ebenfalls durch Prolyl-3-Hydroxylierung modifiziert wird, sind auch die Epiphysen betroffen.
Typ VIII (selten, gehäuft bei irischen Auswanderern und bei Westafrikanern, bei denen 1% der Bevölkerung Anlageträger sind und Typ VIII genauso häufig ist wie OI Typ II) ist gekennzeichnet durch weiße Skleren, einen kurzen fassförmigen Thorax, lange Hände und extrem untermineralisierte Knochen. Ursächlich sind Varianten im Prolyl-3-Hydroxylase-Gen LEPRE1, das Pro986 der alpha1(I)-Kette modifiziert (post-translationale Prolyl-3-Hydroxylierung von Kollagen Typ I und II). Varianten zeigen einen vergleichbaren strukturellen Effekt und führen zu einer vergleichbaren klinischen Symptomatik wie OI Typ VII.
Typ IX (selten) ist eine mäßig schwere Form der OI ohne Rhizomelie. Sie wird verursacht durch Varianten im PPIB-Gen. PPIB codiert eine Peptidyl-Prolyl-cis-trans-Isomerase, die die Prolyl-Isomerisierung katalysiert und für die Faltung des Typ I-Kollagens essenziell ist.
Tab.: Diagnostische Kriterien für Osteogenesis imperfecta (OI)
Merkmal | Typ I | Typ II | Typ III | Typ IV | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Blaue, violette oder graue Skleren | + | + | + | - | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Minderwuchs | + | entfällt | ++ | + | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schwerhörigkeit | ca. 50% der Fälle | entfällt | ++ | - | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Zhytnik et al. 2019, Mol Genet genomic Med 7:e559 / Caparros-Martin et al. 2017, Mol Genet Genomic Med 5:28 / Bardai et al. 2016, Osteoporosis Int 27:3607 / Forlino et al. 2016, Lancet 387:1657 / Valadares et al. 2014, J Pediatr 90:536 / Caparros-Martin et al. 2013, Am J Med Genet 161:1354 / van Dijk et al. 2012, EJHG 20:11 / Forlino et al. 2011, Nat Rev Endocrinol 7:540 / Alanay et al. 2010, Am J Hum Genet 86:551 / Barnes et al. 2010, NEJM 362:521 / Willaert et al. 2009, J Med Genet 46:233 / Baldridge et al. 2008, Hum Mutat 29:1435 / Marini et al. 2007, Hum Mutat 28:209
letzte Aktualisierung: 5.11.2023