Thorakale Aortenerkrankungen (TAAD)
Thorakale Aortenaneurysmen und Dissektionen (TAAD) können genetisch bedingt oder isoliert auftreten, wobei 10-20% autosomal-dominant vererbt werden. Sie sind mit verschiedenen Syndromen wie Marfan-Syndrom, Loeys-Dietz-Syndrom und Ehlers-Danlos-Syndrom verbunden. Für isolierte familiäre TAAD wurden bisher neun Gene identifiziert. Die genetische Diagnostik mittels NGS hilft bei der klinischen Differenzialdiagnose von Aortenerkrankungen.
Wissenschaftlicher Hintergrund
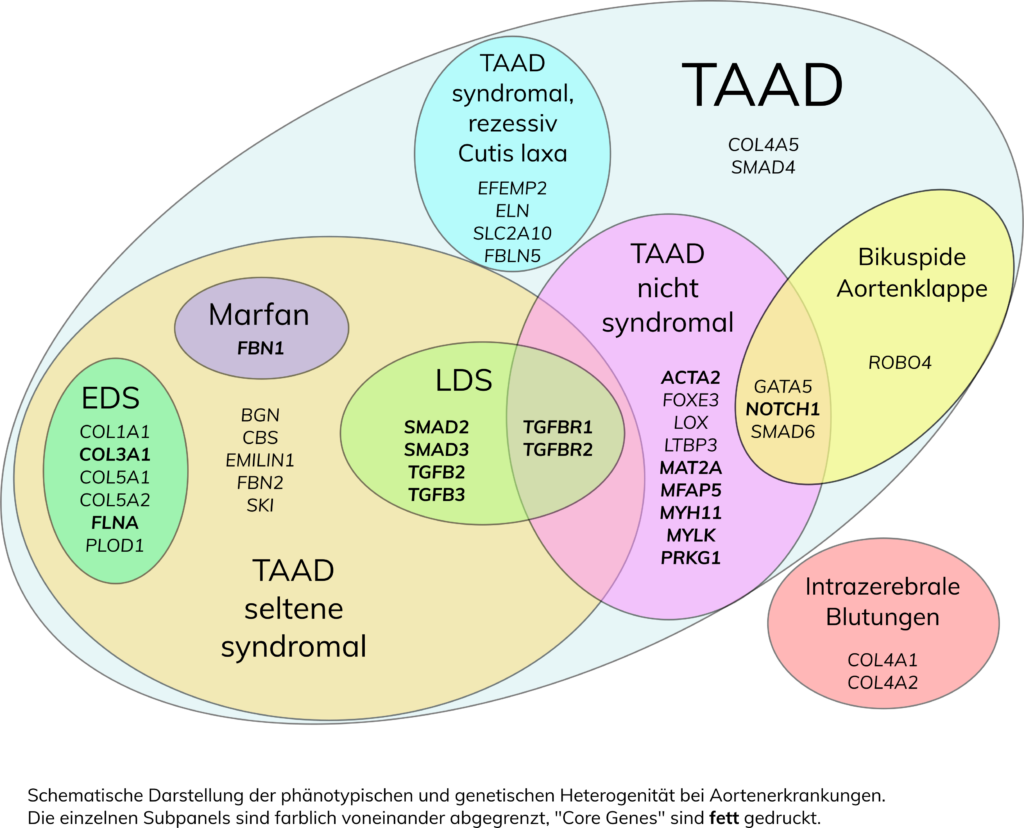
Thorakale Aortenaneurysmen und Dissektionen (TAAD), die die Aorta ascendens unmittelbar hinter der Aortenklappe (Typ A-Dissektion) oder die Aorta descendens im Bereich der linken Arteria subclavia distal des Aortenbogens (Typ B-Dissektion) betreffen, können in Verbindung mit einem genetisch bedingten Syndrom oder isoliert vorkommen. Etwa 10-20% sind autosomal-dominant vererbt, mit reduzierter Penetranz und variabler Expression. TAAD sind sowohl klinisch als auch genetisch heterogen. Zu den syndromalen Aortenerkrankungen zählen:
- Marfan-Syndrom (MFS; FBN1-Gen)
- Loeys-Dietz-Syndrom Typ 1 (LDS1; TGFBR1-Gen)
- Loeys-Dietz-Syndrom Typ 2 (LDS2; TGFBR2-Gen)
- Loeys-Dietz-Syndrom Typ 3 (LDS3; SMAD3-Gen)
- Loeys-Dietz-Syndrom Typ 4 (LDS4; TGFB2-Gen)
- Loeys-Dietz-Syndrom Typ 5 (LDS5; TGFB3-Gen)
- Loeys-Dietz-Syndrom Typ 6 (LDS6; TGFB2-Gen)
- Meester-Loeys-Syndrom (MRLS; BGN-Gen)
- Ehlers-Danlos-Syndrom, vaskulärer Typ (vEDS; COL3A1-Gen)
- Ehlers-Danlos-Syndrom mit periventrikulärer Heterotopie (EDS, PVNH4; FLNA-Gen)
- Ehlers-Danlos-Syndrom, klassischer Typ (cEDS; COL5A1-Gen, COL5A2-Gen, COL1A1-Gen)
- Ehlers-Danlos-Syndrom, kyphoskoliotischer Typ (kEDS; PLOD1– Gen)
- Seltene Syndrome mit Risiko für TAAD: Arterial Tortuosity Syndrom (ATS; SLC2A10-Gen), autosomal dominante und autosomal rezessive Cutis laxa (ADCL1, ADCL2, ARCL1A, ARCL1B, ARCL1C; ELN, FBLN5, EFEMP2, LTBP4) .
Darüber hinaus gibt es auch bei weiteren Bindegewebserkrankungen ein Risiko für Aortenaneurysmen und Dissektionen.
Für isolierte familiäre TAAD wurden in Kopplungsanalysen bisher 11 Genorte lokalisiert und neun Gene identifiziert:
- AAT3 auf Chromosom 3p24-25 (TGFBR2-Gen)
- AAT4 auf Chromosom 16p13.13-p13.12 (MYH11-Gen)
- AAT5 auf Chromosom 9q33-q34 (TGFBR1-Gen)
- AAT6 auf Chromosom 10q22-24 (ACTA2– Gen)
- AAT7 auf Chromosom 3q21 (MYLK-Gen)
- AAT8 auf Chromosom 10q11.2-q21.1 (PRKG1-Gen)
- AAT9 auf Chromosom 12p13.31 (MFAP5-Gen)
- AAT10 auf Chromosom 5q23.1 (LOX-Gen)
- AAT11 auf Chromosom 1p33 (FOXE3-Gen)
Für AAT1 auf Chromosom 11q23-24 und AAT2 auf Chromosom 5q13-14 wurde bisher kein Gen identifiziert. Weitere Gene mit pathogenen Varianten bei TAAD sind MAT2Aund SMAD2, sowie GATA5, NOTCH1, ROBO4und SMAD6, wobei Veränderungen in den vier letztgenannten Ursachen für eine bikuspide Aortenklappe darstellen.
Da die klinische Differenzialdiagnose bei Aortenerkrankungen oft schwierig ist, stellt die genetische Diagnostik mittels NGS eine Möglichkeit der Ursachenfindung dar.
Renard et al. 2018, J Am Coll Cardiol 72:605 / Milewicz and Regalado In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® (Updated 2017 Dec 14) / Arslan-Kirchner et al. 2016, Eur J Hum Genet 24 / Bowdin et al. 2016, Canadian J Cardiol 32:131 / Zhang and Wang 2016, Clin Genet 89:639 / Guo et al. 2013, Am J Hum Genet 93:398 / Boileau et al. 2012, Nat Genet 44:916 / van de Laar et al. 2011, Nat Genet 43:121 / Wang et al. 2010, Am J Hum Genet 87:701 / Guo et al. 2007, Nat Genet 39:1488 / Zhu et al. 2006, Nat Genet 38:343 / Pannu et al. 2005, Circulation 112:513 / Dietz et al. 2005, Am J Med Genet 139C:4
letzte Aktualisierung: 18.3.2024