Marfan Syndrom
Das klassische Marfan-Syndrom (MFS), eine häufige Bindegewebserkrankung, wird durch pathogene Varianten im FBN1-Gen verursacht, das für Fibrillin-1 codiert, einen wesentlichen Bestandteil der extrazellulären Matrix. Diese genetischen Veränderungen können zu einem breiten Spektrum von Symptomen in verschiedenen Organsystemen führen, insbesondere im Herz- und Gefäßsystem, Skelett und Augen. Neben pathogenen Varianten in FBN1 können Varianten in den Genen TGFBR1 und TGFBR2 zu Marfan-Syndrom-ähnlichen Erkrankungen führen.
Wissenschaftlicher Hintergrund
Die häufigste Bindegewebserkrankung ist das klassische Marfan-Syndrom (MFS), das durch pathogene Varianten im FBN1-Gen bedingt ist, welches für Fibrillin-1 codiert. Fibrillin wird von den Fibroblasten sezerniert und ist neben Kollagen und Elastin der wichtigste strukturelle Bestandteil der extrazellulären Bindegewebsmatrix. Durch das weit verbreitete Mikrofibrillensystem im Organismus können pathogene Fibrillin-1-Varianten zu einem breiten Spektrum von klinischen Manifestationen in verschiedenen Organsystemen führen. Bei den klinischen Symptomen stehen die Beteiligung von Herz- und Gefäßsystem, Skelett und die Augen (Linsenluxation) im Vordergrund. Anhand der revidierten Ghenter Nosologie von 2010 kann bei Vorliegen einer isolierten Aortenwurzeldilatation bzw. -dissektion oder einer isolierten Linsenluxation mit dem Nachweis einer pathogenen Variante im FBN1-Gen die Diagnose Marfan-Syndrom gesichert werden, wenn diese im Zusammenhang mit einer Aortenwurzelerweiterung beschrieben ist. Dagegen führt eine Linsenluxation mit dem Nachweis einer pathogenen, heterozygoten FBN1-Variante, die nicht mit Aortenwurzeldilatation assoziiert ist, zur Diagnose eines Ektopia Lentis-Syndroms (ECTOL1). Pathogene FBN1-Varianten sind bei Patienten mit klassischem Marfan-Syndrom beschrieben, aber auch bei den alternativen Diagnosen MASS-Phänotyp (Myopie, Mitralklappenprolaps, grenzwertige Aortenwurzeldilatation, Striae und Skelettbeteiligung) und Mitralklappenprolaps-Syndrom (MVPS). Bei Vorliegen einer Aortenwurzeldilatation (Z>2) und negativer FBN1-Analyse wird eine Untersuchung der Gene TGFBR1 und TGFBR2 empfohlen.
Das FBN1-Gen erstreckt sich über 230 Kilobasen genomischer DNA und besteht aus codierenden 66 Exons. Substitutionen eines der sechs konservierten Cysteinreste sind die häufigsten Aminosäureaustausche innerhalb der 43 Kalzium-bindenden (cb), Epidermal Growth Factor (EGF)-ähnlichen Domänen. Sie führen zum Verlust einer Disulfid-Brücke und beinträchtigen somit die Faltung von Fibrillin-1. Die Konsequenz ist ein reduzierter Einbau intakter Mikrofibrillen in die extrazelluläre Matrix durch einen dominant negativen Effekt der veränderten Moleküle.
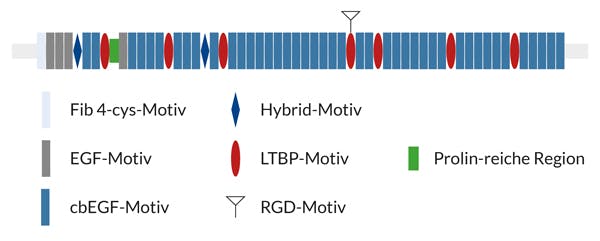
Abb.: Schematische Darstellung der verschiedenen Domänen des Fibrillin-1-Proteins: 47 EGF-Motive, von denen 43 zusätzlich eine Ca2+-bindende Konsensussequenz aufweisen (cbEGF). In Wiederholungen angeordnete cbEGF-Motive sind durch LTBP-Motive, die Homologie zum Latent Transforming Growth Factor ß Binding Protein aufweisen, und Hybrid-Motive zwischen cbEGF und LTBP unterbrochen
Bei Patienten mit klassischem Marfan-Syndrom, bei denen die klinische Diagnose anhand der Ghenter Kriterien von 1996 gestellt werden kann, werden in bis zu 95% pathogene Varianten im FBN1-Gen identifiziert. Bei Patienten, die eine Teilsymptomatik eines MFS mit zusätzlichen Merkmalen zeigen (Patienten mit Marfan-ähnlichem Syndrom oder unvollständiger Marfan-Symptomatik), und bei denen keine pathogene Variante im FBN1-Gen nachgewiesen werden kann, konnten in 5-25% Varianten in den Genen für die Transforming Growth Factor ß Rezeptoren 1 und 2 (TGFBR1 und TGFBR2) identifiziert werden. Die Mutationserfassungsrate in TGFBR1 und TGFBR2 bei thorakalen Aortenaneurysmen und Dissektionen (TAAD) liegt bei etwa 5%.
Muiño-Mosquera et al. 2018, Circ Genom Precis Med 11:e002039 / Groth et al. 2017, Genet Med 19:772 / Dietz in Pagon RA, Adam MP, Ardinger HH, et al. (eds). GeneReviews® (Updated 2017 Oct 12) / Verstraeten et al. 2016, Hum Mutat 37:524 / De Backer et al. 2015, Curr Pharm Des 21:4061 / von Kodolitsch et al. 2015, Appl Clin Genet / Loeys et al. 2010, J Med Genet 47:476 / Stheneur et al. 2008, Hum Mutat 29:E284 / Faivre et al. 2007, Am J Hum Genet 81:454 / Collod-Béroud and Boileau 2002, Eur J Hum Genet 10:673
letzte Aktualisierung: 9.4.2024